Abstract
Experimental autoimmune encephalomyelitis (EAE) is a well-established animal model of human multiple sclerosis (MS). We have evaluated the role of oxidative and nitrosative stress, as the causal factors in the development of EAE, responsible for the damage of cardinal cellular components, such as lipids, proteins and nucleic acids, resulting in demyelination, axonal damage, and neuronal death. EAE was induced in female Sprague-Dawley rats, 3 months old (300 ± 20 g), by immunization with myelin basic protein in combination with Complete Freund's adjuvant (CFA). The animals were divided into seven groups: control, EAE, CFA, EAE + aminoguanidine (AG), AG, EAE + N-acetyl-l-cysteine (NAC) and NAC. The animals were sacrificed 15 days after EAE induction, and the levels of nitrosative and oxidative stress were determined in 10% homogenate of the whole encephalitic mass. In EAE rats, brain NO production and MDA level were significantly increased (P < 0.001) compared to the control values, whereas AG and NAC treatment decreased both parameters in EAE rats compared to EAE group (P < 0.001). Glutathione (GSH) was reduced (P < 0.001) in EAE rats in comparison with the control and CFA groups, but increased in EAE + AG and EAE + NAC group compared to the EAE group (P < 0.01). Superoxide dismutase (SOD) activity was significantly decreased (P < 0.001) in the EAE group compared to all other experimental groups. The clinical expression of EAE was significantly decreased (P < 0.05) in the EAE groups treated with AG and NAC compared to EAE rats, during disease development.
The obtained results prove an important role of oxidative and nitrosative stress in the pathogenesis of EAE, whereas AG and NAC protective effects offer new possibilities for a modified combined approach in MS therapy.
Introduction
Experimental allergic encephalomyelitis (EAE) is an inflammatory demyelinating disease of the central nervous system (CNS) with clinic-pathological and immunological similarities to multiple sclerosis.Citation1 This experimental disease can be induced in susceptible animals by active immunization with myelin basic protein in an adequate strong adjuvant. Multiple sclerosis (MS) is a chronic inflammatory, demyelinating disease of CNS which is characterized by neural tissue damage. The recent data of MS suggest biphasic MS pathomechanism based on demyelination (relapsing remitting (RR) phase) and neurodegeneration (progressive phase). Inflammatory cascades, as microglial activation consequences, predominate in the RR phase, whereas oxidative stress and excitotoxicity prevail during the progressive phase. In both cases, inflammatory and degenerative cascades are interactive.Citation2–Citation4
In physiological conditions, the basal levels of reactive oxygen species (ROS) are generated due to normal metabolism processes. Normal ROS concentrations are preserved by antioxidant enzymes (superoxide dismutase (SOD) catalase) and non-enzymatic low molecular antioxidants (uric acid, ascorbic acid). The elevated levels of ROS during pathological conditions, produced by inflammatory processes and mitochondria respiratory chain dysfunction, lead to oxidative stress. The most important effector molecule in the progress of oxidative stress and its damage is peroxynitrite (ONOO−), formed by the reaction of nitric oxide (NO•) with superoxide (O2−). Nitric oxide is an essential biological molecule produced by nitric oxide synthase (nNOS) in neurons and endothelial cells (eNOS) during physiological conditions, or in mitochondria and microglia by inducible nitric oxide synthase (iNOS) during inflammatory processes. The high NO• level leads to the rapid reaction with superoxide to form peroxynitrite, or with other biomolecules (proteins, DNA, lipids).Citation5,Citation6 Peroxynitrite, playing an important role in neuronal tissue damage, induces mitochondrial dysfunction, lipid peroxidation, protein nitration, ion channel disability and electrolyte dysbalance.Citation7–Citation9
In an attempt to elucidate the role of NO in the development of EAE, several studies have employed iNOS inhibitors to treat EAE. One of these substances is aminoguanidine (AG), an analog of l-arginine. AG is equipotent to NG-monomethyl-l-arginine (l-NMA), an inhibitor inducible isoform of NO synthase, but 10–100-fold less potent as an inhibitor of the constitutive isoforms,Citation10 which makes this drug a useful selective inhibitor of iNOS. While several investigators have reported that AG inhibits the clinical signs of EAE, the others have found the aggravation and prolongation of the disease upon AG treatment.Citation11,Citation12
Considering the associated oxidative stress in EAE, the literature data point out the possible protective role of an oxidant-scavenger, N-acetyl-l-cysteine (NAC), which has been shown to be beneficial against ROS generation, decrease mitochondria-related oxidative stress and protect mitochondria from damage in CNS.Citation13
On the other hand, an imbalance between excitatory and inhibitory neurotransmission, due to glutamate overload, leading to an excessive generation of excitatory amino acids, could also induce neuronal damage.Citation14 Normally, astrocytes take up glutamate from extracellular spaces and convert it to glutamine. It is released to extracellular space by astrocytes, taken up by neurons and converted back to glutamate. The mechanisms of excitotoxicity are caused by channellopathy, calcium overload, mitochondriopathy, proteolytic enzyme production and the activation of the pathways leading to cell damage.Citation15,Citation16
In this work we investigate potential nitrosative and oxidative stress modulators and estimate their protective effects and new possibilities for a modified combined approach in MS therapy.
Materials and methods
Animals
Female Sprague-Dawley rats, 3 months old, weighing 300 ± 20 g, were housed in the Biomedical Research Centre animal care facility of the Medical Faculty of Nis throughout the experiment under a 12:12 hours light–dark cycle. The rats were kept in plastic cages and fed on a standard diet and water ad libitum. All animals received human care in the strict accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publication 80–23, revised 1985). The experimental protocol was reviewed and approved by the Faculty Ethical Committee.
Induction of EAE
EAE was induced by the subcutaneous injection of myelin basic protein, bovine type (50 µg), dissolved in phosphate buffered saline (PBS) emulsified in equal volume of the complete Freund's adjuvant (CFA), on days 0 and 7 in the hind foot pad of the animals under anesthesia. Two intraperitoneal injections of 200 ng Pertussis toxin were given on days 0 and 1. Each of 49 animals, randomly, was assigned to seven groups: control (PBS 0.3 ml/i.p/daily), EAE (PBS 0.3 ml/i.p/daily after EAE induction), CFA (0.3 ml/i.p/daily), EAE and AG (AG 100 mg/kg body weight/daily after EAE induction), AG (100 mg/kg body weight/daily), EAE and NAC (150 mg/kg body weight/daily after EAE induction), and NAC (100 mg/kg body weight/daily).
All animals were scored daily, for the clinical signs of EAE (healthy = 1; loss of tail tone = 2; hindlimb weakness = 3; hindlimb paralysis = 4; hindlimb paralysis plus forelimb weakness = 5; moribund or dead = 6). The animals were sacrificed 15 days after EAE induction and the brains were dissected, washed in PBS, placed on ice, and 10% homogenates of the whole encephalitic mass (WEM) were stored at −20°C for later biochemical analysis. Nitrite and nitrate concentration, malonyl dialdehide (MDA) and glutathione (GSH) levels and SOD activity were determined.
Determination of nitrate and nitrate concentration
After deproteinization, the production of NO• was evaluated by measuring nitrite and nitrate concentrations. Nitrites were assayed directly spectrophotometrically at 543 nm, using the colorimetric method of Griess (Griess reagent: 1.5% sulfanilamide in 1 M HCl plus 0.15% N-(1-naphthyl)ethylendiamine dihydrochloride in distilled water). However, nitrates were previously transformed into nitrites by cadmium reduction.Citation17
Determination of malondialdehyde
The lipid peroxidation level was measured spectrophotometrically by the estimation of malondialdehyde concentration (nmol/mg of tissue weight) based on the reaction with thiobarbituric acid.Citation18
Reduced GSH concentration
Reduced GSH concentration (nmol/mg of tissue weight) was measured using Elman's reagent.Citation19
SOD activity
SOD activity was measured by the method of Minami and Yoshikawa, based on the inhibition of pyrogallol autooxidation by plasma SOD.Citation20 Superoxide anion, formed by pyrogallol autooxidation, forms a colored product in the reaction with NBT. As O2− scavenger, SOD inhibits this reaction, while one unit of the ezyme activity represents 50% inhibition.
Protein content
Protein content was measured according to the Lowry procedure using bovine serum albumin as standard.Citation21
Chemicals
Chemicals were purchased from Sigma (St Louis, MO, USA). All used chemicals were of analytical grade. All drug solutions were prepared on the day of the experiment.
Statistical analysis
All the data presented were mean ± SD. Normal distribution was verified using Kolmogorov–Smirnov test. The data were analyzed statistically by one-way ANOVA and Chi-square test, using the statistical program SPSS version 13. Statistical significance was defined as P < 0.05.
Results
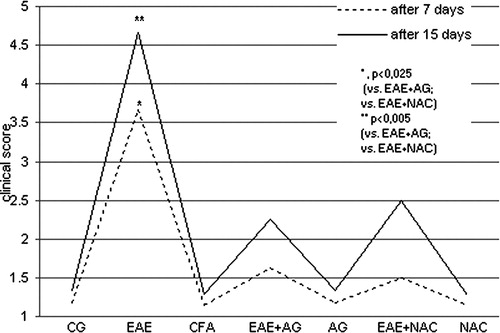
The clinical expression of EAE was significantly decreased in the EAE groups treated with AG and NAC during disease development compared to EAE group (P < 0.025, after 7 days, P < 0.05 after 15 days; ).
Figure 1. The clinical score of neurological disorder (1–6) of rats after 7 and 15 days. CG – control group; EAE – rats with experimental autoimmune encephalomyelitis; CFA – rats treated with complete Freund's adjuvant; EAE + AG – EAE rats treated with aminoguanidine; AG – rats treated with aminoguanidine; EAE + NAC – EAE rats treated with N-acetyl-l-cysteine; NAC – rats treated with N-acetyl-l-cysteine. 1 – healthy rats; 2 – loss of tail tone; 3 – hindlimb weakness; 4 – hindlimb paralysis; 5 – hindlimb paralysis plus forelimb weakness; 6 – moribund or dead.
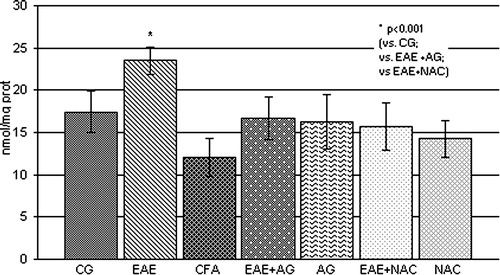
The obtained results showed that the level of nitrate and nitrite, as a measure of NO production, was significantly increased in EAE rats compared to the control and CFA groups (P < 0.001; ), Also, the brain concentration of MDA, as a final lipid peroxidation product, was significantly increased compared to control values (P < 0.001; ), whereas AG and NAC treatment decreased both parameters in EAE rats compared to the EAE group (P < 0.001; and ).
Figure 2. NO2 and NO3 concentration (nmol/mg prot.) in the rat WEM. CG – control group; EAE – rats with experimental autoimmune encephalomyelitis; CFA – rats treated with complete Freund's adjuvant; EAE + AG – EAE rats treated with aminoguanidine; AG – rats treated with aminoguanidine; EAE + NAC – EAE rats treated with N-acetyl-l-cysteine; NAC – rats treated with N-acetyl-l-cysteine.
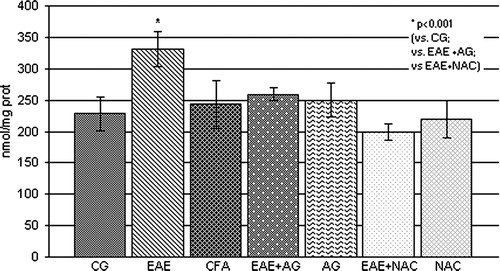
Figure 3. MDA concentration (nmol/mg prot.) in the rat WEM. CG – control group; EAE – rats with experimental autoimmune encephalomyelitis; CFA – rats treated with complete Freund's adjuvant; EAE + AG – EAE rats treated with aminoguanidine; AG – rats treated with aminoguanidine; EAE + NAC – EAE rats treated with N-acetyl-l-cysteine; NAC – rats treated with N-acetyl-l-cysteine.
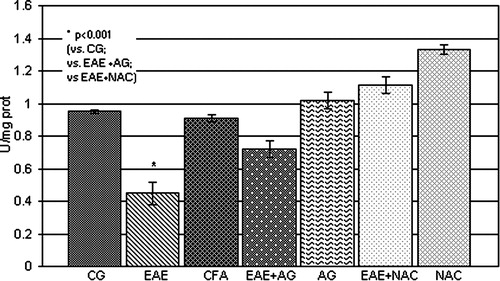
The GSH level and SOD activity were significantly reduced (P < 0.001; and , respectively) in EAE rats in comparison with the control and CFA group. The treatment of EAE rats with AG and NAC significantly increased both parameters compared to EAE (P < 0.01; and ).
Figure 4. GSH concentration (nmol/mg prot.) in the rat WEM. CG – control group; EAE – rats with experimental autoimmune encephalomyelitis; CFA – rats treated with complete Freund's adjuvant; EAE + AG – EAE rats treated with aminoguanidine; AG – rats treated with aminoguanidine; EAE + NAC – EAE rats treated with N-acetyl-l-cysteine; NAC – rats treated with N-acetyl-l-cysteine.
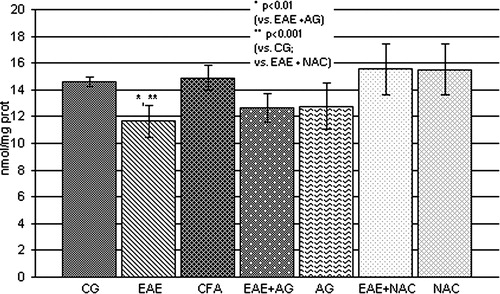
Figure 5. SOD activity (U/mg prot.) in the rat WEM. CG – control group; EAE – rats with experimental autoimmune encephalomyelitis; CFA – rats treated with complete Freund's adjuvant; EAE + AG – EAE rats treated with aminoguanidine; AG – rats treated with aminoguanidine; EAE + NAC – EAE rats treated with N-acetyl-l-cysteine; NAC – rats treated with N-acetyl-l-cysteine.
Discussion
MS has long been considered as an immune-mediated inflammatory disease. It was generally believed that demyelination caused by the inflammatory process was responsible for both neurological deficits and disability progression. An accumulating body of evidence has challenged this earlier concept, demonstrating that disability results from neuronal axonal loss rather than demyelination. The pathomechanisms of neurodegeneration are basically mediated by oxidative stress and excitotoxicity. Inflammation, which is the base of the early EAE stage, might raise ROS levels leading to oxidative stress.Citation22 Oxygen and nitrogen free radicals generated by macrophages have been implicated as mediators of demyelination and axonal injury in both EAE and MS.Citation22,Citation23 Free radicals can activate certain transcription factors, such as nuclear transcription factor-kappa B (NF-κB), which upregulates the expression of many genes involved in EAE, such as tumor necrosis factor-α (TNF-α), iNOS, and other biomolecules.Citation24 Clinical trials of patients with MS have shown increased free radical activity, with contradictory reports on important antioxidant enzyme activities compared with healthy controls.Citation25
The reactions, initiated by the reactive species, include peroxidation of lipids, nitrosylation of thiol groups, nitration of tyrosine, and the oxidation and deamination of nucleic acids. Several investigators have shown that the inhibition of the inducible/inflammatory form of NOS as well as the use of the anti-sense knockdown of iNOS suppress EAE in mice and rats.Citation26 Also, Hooper et al.Citation26 demonstrated that the inhibition of iNOS, or the scavenging of NO• or peroxynitrite by antioxidants, inhibited neurological deficits in mice with EAE. Meanwhile, the therapeutic modulation of NO• precursors, such as inducible NOS, nitric oxide or superoxide, appeared delicate and yielded only modest and contradictory results in EAE.Citation27,Citation28 Our results show that the maximum clinical severity of EAE and the duration of illness were significantly reduced by the application of AG (). There are two possible mechanisms to explain this protective effect of AG. As a highly reactive nucleophilic reagent that reacts with many biological molecules (pyridoxal phosphate, pyruvate, glucose, malonyl dialdehyde, and others), AG, NOS inhibitor, preferentially inhibits the cytokine and endotoxin inducible isoform of NOS versus the constitutive isoforms. Since iNOS-positive cells and/or nitrotyrosine-positive cells are found in close proximity to blood vessels, especially during the early phase of EAE,Citation11,Citation29 NO• from these cells may contribute to destruction of the blood–brain barrier. The inhibition of NO• production might block infiltration of inflammatory cells into the CNS. Second, since NO• is cytotoxic and is considered to cause tissue injury in the CNS,Citation30 the overproduction of NO• can expand inflammation. The inhibition of NO• release might prevent such tissue injury and, as a result, might suppress inflammation in the CNS, during the early phase of EAE.
In EAE, peroxynitrite is formed very early, correlates with disease activity and cannot be detected during remissions and chronic, silent stages.Citation31 Some studies have also shown that nitrite and nitrate (metabolites of NO•) are significantly elevated in MS patients, and that the levels are directly related to the disease state.Citation32 Early reports focused on the possibility that increased NO• production, induced by proinflammatory cytokines interleukin (IL-1) and interferon-gamma (INF-gamma), was responsible for tissue injury.Citation31 This possibility is based on the observations that reactive nitrogen species (RNS) are toxic to oligodendrocytes, they stimulate axon demyelization, and also that NO• inhibitors are protective against EAE development.Citation33 But, the recent findings have questioned the purely destructive role of NO•. In particular, the targeted deletion of the iNOS gene in mice enhanced their susceptibility to EAE and also specific iNOS inhibition was found to prolong the disease, supporting a putative role of NO• as an immunosuppressive and anti-inflammatory molecule.Citation34
An additional study showed that the administration of NAC, which can effectively raise intracellular GSH levels, reduced the intensity of acute EAE clinical signs, which was proved in our study ( and ). The protection was associated with the enhancement of the specific lymphocyte proliferative response to the immunizing antigens at early stages, post-encephalitogenic injection.Citation35,Citation36 Antioxidant NAC is able to protect cultured oligodendrocytes from toxicity mediated by the proinflammatory cytokine TNF-α.Citation36 The oral administration of NAC substantially inhibited the development of EAE in mice, and a recent publication demonstrates a reduction in the cellular infiltrate in the CNS.Citation36 Cell culture studies showed that NAC also inhibited induction of iNOS and NO• production in peritoneal macrophages, C6 glial cells and primary astrocytes, and blocked the activation of NF-κB in peritoneal macrophages. Accordingly, the oral administration of this oxidant scavenger was found to attenuate EAE clinical signs.Citation37,Citation38 These NAC effects are based on the following observations: the NAC treatment of EAE rats (Citation1) reduced the severity of EAE clinical symptoms, (Citation2) attenuated the infiltration of mononuclear cells into the CNS of EAE rats, (Citation3) blocked the induction of proinflammatory cytokines, iNOS, and nitrotyrosine in the CNS, and (Citation4) decreased proinflammatory Th1 cytokine responses (IFN-gamma) from ex vivo splenocytes, while increasing anti-inflammatory cytokine production (IL-10), and decreasing NO• production in lipopolysaccharide (LPS)-stimulated splenocytes.Citation38,Citation39
The increased levels of the secondary products of oxidative stress and/or the decreased levels of antioxidant enzymes and small molecule antioxidants are seen in blood and CSF during the active phases of MS.Citation40 Independent of inflammation, evidence for mechanisms leading to neuronal degeneration in MS include mitochondrial dysfunction and an excitotoxic component. In EAE, the nitration of mitochondrial proteins, which preceded the infiltration of inflammatory cells, resulted in the loss of mitochondrial membrane potential and apoptotic cell death.Citation40,Citation41
NO• can decrease the activity of some of the enzymes involved in antioxidant defense in cultured oligodendrocyte like cells. Thus, it was found to decrease the activities of the antioxidant enzymes GSH peroxidase, catalase, and Mn-SOD, probably through the down-regulation of the expression of their mRNAs. Such observations indicate that NO• production may affect the level of oxidative stress and the effects of ROS production.Citation42 There are three forms of SOD: Mn-SOD, located in mitochondria; Cu/Zn-SOD, located within the cytosol; and SOD3 is the only enzyme that scavenges superoxide in extracellular compartment and limits the formation of strong neurotoxic oxidants including hydroxyl radical and peroxynitrite.Citation43 It has been demonstrated that SOD3 can attenuate tissue damage and inflammation.Citation44 Together with the reduced GSH content, decreased SOD activity in our experiment proves reduced antioxidant defense and offers a possibility of increased peroxynitrite formation in these conditions. Attempts to increase SOD activity could be a potentially valuable tool and of relevance to treatment strategies for the inflammatory diseases of CNS.
These results prove an important role of oxidative and nitrosative stress in the pathogenesis of EAE, whereas AG and NAC protective effects offer new possibilities for a modified combined approach in MS therapy.
Acknowledgments
Supported by the Serbian Ministry of Science and Technological Development (project number 41018).
References
- Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol 2007;7:904–12.
- Gonsette RE. Oxidative stress and excitotoxicity: a therapeutic issue in multiple sclerosis? Mult Scler 2008;14:22–34.
- Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res 2005;81:374–89.
- Jack C, Ruffini F, Bar-Or A, Antel JP. Microglia and multiple sclerosis. J Neurosci Res 2005;81:363–73.
- Bast A, Haenen GRMM, Doelman CJA. Oxidants and antioxidants: state of the art. Am J Med 1991;91:2S–13S.
- Beckman JS, Chen J, Crow JP, Ye YZ. Reactions of nitric oxide, superoxide and peroxynitrite with superoxide dismutase in neurodegeneration. Prog Brain Res 1994;103:371–80.
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 1996;271:C1424–37.
- Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide 2010;23(2):75–93.
- Marques C, Cheeran M, Palmquist J, Hu S, Lokensgard J. Microglia are the major cellular source of inducible nitric oxide synthase during experimental herpes encephalitis. J Neurovirol 2008;14(3):229–38.
- Misko TP, Moore WM, Kasten TP, Nickols GA, Corbett JA, Tilton RG, et al. Selective inhibition of the inducible nitric oxide synthase by amino- control inflammation in the brain. Eur J Pharmacol 1993;233:119–25.
- Brenner T, Brocke S, Szafer F, Sobel RA, Parkinson JF, Perez DH, et al. Inhibition of nitric oxide synthase for treatment of experimental autoimmune encephalomyelitis. J Immunol 1997;158:2940–6.
- Ruuls SR, Van der Linden S, Sontrop K, Huitinga I, Dijkstra CD. Aggravation of experimental allergic encephalomyelitis (EAE) by administration of nitric oxide (NO) synthase inhibitors. Clin Exp Immunol 1996;103:467–74.
- Kamboj SS, Sandhir R. Protective effect of N-acetylcysteine supplementation on mitochondrial oxidative stress and mitochondrial enzymes in cerebral cortex of streptozotocin-treated diabetic rats. Mitochondrion 2011;11(1):214–22, 7.
- Zaffaroni M. Biological indicators of the neurodegenerative phase of multiple sclerosis. Neurol Sci 2003;24:279–82.
- Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol 1996;9:836–44.
- Napoli I, Neumann H. Protective effects of microglia in multiple sclerosis. Exp Neurol 2010;225(1):24–8.
- Navaro-Gonzalvez JA, Garcia-Benayas C, Arenas J. Semiautomated measurement of nitrate in biological fluids. Clin Chem 1998;44:679–81.
- Andreeva LI, Kozhemiakin LA, Kishkun AA. Modification of the method of determining lipid peroxidation in a test using thiobarbituric acid. Lab Delo 1988;11:41–3.
- Sedlak J, Lindsday R. Estimation of total protein bound and non-protein sulphydryl groups in tissue with Ellman's reagent. Anal Biochem 1968;25:192–205.
- Minami M, Yoshikawa H. A simplified assay method of superoxide dismutase activity for clinical use. Clin Chim Acta 1979;92:337–42.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with folin phenol reagent. J Biol Chem 1951;193:265–75.
- Leung G, Sun W, Zheng L, Brookes S, Tully M, Shi R. Anti-acrolein treatment improves behavioral outcome and alleviates myelin damage in experimental autoimmune enchephalomyelitis mouse. Neuroscience 2011;173:150–5.
- Henderson A, Barnett M, Parratt J, Prineas J. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol 2009;66(6):739–53.
- Glass C, Saijo K, Winner B, Marchetto M, Gage F. Mechanisms underlying inflammation in neurodegeneration. Cell 2010;140(6):918–34.
- Bishop A, Hobbs K, Eguchi A, Jeffrey S, Smallwood L, Pennie C, et al. Differential sensitivity of oligodendrocytes and motor neurons to reactive nitrogen species: implications for multiple sclerosis. J Neurochem 2009;109(1):93–104.
- Hooper DC, Spitsin S, Kean RB, Champion JM, Dickson GM, Chaudhry I, et al. Uric acid, a natural scavenger of peroxynitrite in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci USA 1998;95:675–80.
- Van der Veen RC, Roberts LJ. Contrasting roles for nitric oxide and peroxynitrite in the peroxidation of myelin lipids. J Neuroimmunol 1999;95:1–7.
- Gonsette RE. Neurodegeneration in multiple sclerosis: the role of oxidative stress and excitotoxicity. J Neurol Sci 2008;274:48–53.
- Van Dam AM, Bauer J, Man-A-Hing WKH, Marquette C, Tilders FJH, Berkenbosch F. Appearance of inducible nitric oxide synthase in the rat central nervous system after rabies virus infection and during experimental allergic encephalomyelitis but not after peripheral administration of endotoxin. J Neurosci Res 1995;40:251–60.
- Sherman MP, Griscavage JM, Ignarro LJ. Nitric oxide-mediated neuronal injury in multiple sclerosis. Med Hypotheses 1992;39:143–6.
- Van der Veen RC, Hinton DR, Incardonna F, Hofman FM. Extensive peroxynitrite activity during progressive stages of central nervous system inflammation. J Neuroimmunol 1997;77:1–7.
- Yamashita T, Ando Y, Obayashi K, Uchino M, Ando M. Changes in nitrite and nitrate (NO2/-/NO3/-) levels in cerebrospinal fluid of patients with multiple sclerosis. J Neurol Sci 1997;153:32–4.
- Bishop A, Hobbs K, Eguchi A, Jeffrey S, Smallwood L, Pennie C, et al. Differential sensitivity of oligodendrocytes and motor neurons to reactive nitrogen species: implications for multiple sclerosis. J Neurochem 2009;109(1):93–104.
- Arnett HA, Hellendall RP, Matsushima GK, Suzuki K, Laubach VE, Sherman P, et al. The protective role of nitric oxide in a neurotoxicant-induced demyelinating model. J Immunol 2002;168:427–33.
- Lehmann D, Karussis D, Misrachi KR, Shezen E, Ovadia H, Abramsky O. Oral administration of the oxidant-scavenger N-acetyl-L-cysteine inhibits acute experimental autoimmune encephalomyelitis. J Neuroimmunol 1994;50:35–42.
- Stanislaus R, Gilg AG, Singh AK, Singh I. N-acetyl-L-cysteine ameliorates the inflammatory disease process in experimental autoimmune encephalomyelitis in Lewis rats. J Autoimmune Dis 2005;2(1):4.
- Pahan K, Sheikh FG, Namboodiri AM, Singh I. N-acetyl cysteine inhibits induction of NO production by endotoxin or cytokine stimulated rat peritoneal macrophages, C6 glial cells and astrocytes. Free Radic Biol Med 1998;24(1):39–48.
- Malabendu J, Kalipada P. Redox regulation of cytokine-mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radic Biol Med 2005;39:823–31.
- Ferretti G, Bacchetti T, Principi F, Di Ludovico F, Viti B, Angeleri VA, et al. Increased levels of lipid hydroperoxides in plasma of patients with multiple sclerosis: a relationship with paraoxonase activity. Mult Scler 2005;11:677–82.
- Koch M, Ramsaransing GS, Arutjunyan AV, Stepanov M, Teelken A, Heersema DJ, et al. Oxidative stress in serum and peripheral blood leukocytes in patients with different disease courses of multiple sclerosis. J Neurol 2006;253:483–7.
- Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Mitochondrial protein nitration primes neurodegeneration in experimental autoimmune encephalomyelitis. J Biol Chem 2006;281:31950–62.
- Dobashi K, Pahan K, Chahal A, Singh I. Modulation of endogenous antioxidant enzymes by nitric oxide in rat C6 glial cells. J Neurochem 1997;68:1896–903.
- Nozik-Grayck E, Suliman HB, Piantadosi CA. Extracellular superoxide dismutase. Int J Biochem Cell Biol 2005;37(12):2466–71.
- Laurila JP, Laatikainen LE, Castellone MD, Laukkanen MO. SOD3 reduces inflammatory cell migration by regulating adhesion molecule and cytokine expression. PLoS One 2009;4(6):1–8.