Abstract
Besides the fact that prolonged whole-body ischemia causes tissue and organ injury during cardiac arrest, additional damage occurs after the restoration of spontaneous circulation, during which the reperfusion activates a host of intracellular responses. These responses may lead to an increased threshold of oxidant-mediated injury and redox-mediated programed cell death in the stunned myocardium. The aim of this article is to summarize the major intracellular responses occurring from the onset of cardiac arrest until the post-resuscitation period that may lead to redox-mediated programed death of myocardial cells.
Introduction
Cardiac arrest is a daunting medical emergency. Characteristically, it has been found that out of every 100 cardiac arrest victims, 30 will achieve restoration of spontaneous circulation (ROSC), but only 5 will survive the post-resuscitation period.Citation1 Despite recent advances, post-cardiac arrest syndrome remains the main reason for the poor survival of patients who achieve ROSC.
Post-resuscitation myocardial stunning, a component of this syndrome, is the mechanical dysfunction that persists after the ROSC and is characterized by the absence of irreversible damage, as well as by normal or near-normal coronary flow.Citation2 Although various factors contribute to its emergence, ischemia/reperfusion (I/R) has a central role in the pathogenesis of this phenomenon.
Besides the fact that prolonged whole-body ischemia causes global tissue and organ injury during cardiac arrest, additional damage occurs during cardiopulmonary resuscitation (CPR) (partial-flow reperfusion) and especially after ROSC, a period in which the whole-body I/R activates a host of cellular stress responses. The aim of this review is to summarize the major pathophysiological responses occurring from the onset of cardiac arrest until the post-resuscitation period that may lead to redox-mediated programed death of myocardial cells.
Cardiac arrest interval
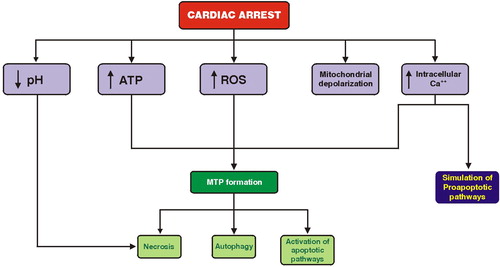
After the onset of cardiac arrest, the intracellular oxygen tension (PO2), carbon dioxide tension (PCO2), and pH change due to the shortage of oxygen. Studies have found that following the onset of ventricular fibrillation, intramyocardial PO2 levels rapidly decrease, whereas PCO2 and hydrogen ion concentration increase.Citation3,Citation4 In addition, mitochondrial oxidative phosphorylation stops and adenosine triphosphate (ATP) is depleted, whereas reactive oxygen species (ROS) are formed and inactivate metabolic enzymes, exacerbating the depletion of myocardial energy.Citation5 As a result, mitochondria depolarize,Citation6 whereas the breakdown of creatine phosphate and ATP increases the intracellular concentration of P+.Citation7
At the same time, glycolysis is accelerated under anaerobic conditions and the concentration of pyruvate, hydrogen ions, and lactate increase.Citation2 Although pyruvate increases cytosolic energy and functions as an antioxidant,Citation8 the prolongation of ischemia will further decrease the intracellular pH. In addition, the cytosolic Ca2+ increases and activates various enzymes, causing alterations in contractile proteins and phospholipid degradation.Citation9 Disturbances in Ca2+ homeostasis and other stresses, such as inhibition of glycosylation and oxidative stress, trigger the endoplasmic reticulum response, which involves the release of signaling proteins and consists of multiple parallel events, such as the apoptotic pathways of stress-activated protein kinases (SAPKs) 1 and 2.Citation10,Citation11
Although the aforementioned factors have a central role in the pathophysiology of post-resuscitation myocardial stunning, the most important intracellular response during this interval is the formation of the large transition pore across the inner and outer mitochondrial membrane. This pore is formed due to the combined effect of oxidative stress, ATP depletion, and sarcolemmal Ca2+ increase.Citation2 The formation of the mitochondrial transition pore (MTP), as well as the mitochondrial ATP-sensitive K+ channel which opens in response to an influx of K+, facilitate the influx of water in mitochondria, thereby countering the effect of matrix shrinkage which is deleterious to respiration.Citation12 However, MTP opening can lead to necrosis through ATP depletion or to apoptosis via cytochrome c release.Citation13 In addition, MTP pore formation can promote autophagy, a regulated process by which a cell carries out lysosomal degradation of its own constituents ().Citation13
Figure 1. Activation of programed cell-death pathways during the cardiac arrest interval.
Redox-mediated responses during CPR
The partial reperfusion of the arrested myocardium and the increased ROS generation by various oxidase systems cause significant changes in oxidant stress and redox state that activate a number of cellular stress responses. In addition, the stunned myocardium is characterized by significant amounts of nitric oxide (NO) which have been generated shortly after the onset of cardiac arrest. Although NO may exert highly protective effects,Citation14,Citation15 it can also play a deleterious role in I/R injury.Citation16 During the CPR interval, the mechanism of mitochondrial oxygen sensing involves redox regulation of NO homeostasis in the inner mitochondrial membrane,Citation17 inhibition of cytochrome c oxidase by NO, and superoxide generation,Citation18 the release of which in the cytosol and the extracellular space is controlled by the mitochondrial membrane and cell membrane anion channels.Citation19 However, NO can interact with superoxide to form peroxynitrite, exacerbating oxidative stress during this interval.
Post-resuscitation period
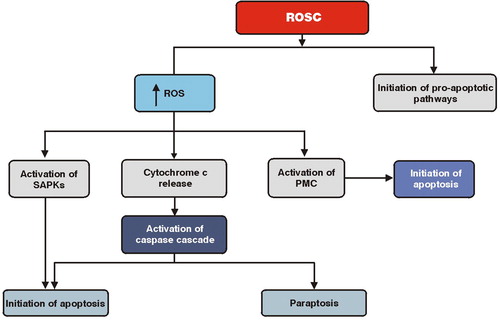
After ROSC, I/R is associated with disturbances in metabolic activity, increases in the production of ROS, and various pro-apoptotic pathways.Citation20–Citation22 Two studies have found that I/R triggers apoptotic cell death through the Fas/Fas ligand pathway and caspase activation.Citation23,Citation24 Several different caspases are activated in I/R-induced apoptosis via two major pathways, the ‘extrinsic’ or death receptor-mediated pathway, which is mediated by specific membrane receptors, such as the tumor necrosis factor and Fas receptors, and the ‘intrinsic’ or mitochondria-mediated pathway, which plays a critical role in the early stages of I/R injury.Citation25–Citation27
In addition, after ROSC and a massive burst of ROS production, the already depolarized mitochondria release cytochrome c, which has a central role not only in the activation of caspase cascade, but also in the permeabilization of the outer mitochondrial membrane.Citation28,Citation29 This permeabilization is regulated by the Bcl-2 family proteins, which interact with the outer mitochondrial membrane and disrupt the normal mitochondrial respiratory function.Citation30 The permeabilized mitochondria, in turn, release the AIF and Endo G factors that translocate to the nucleus and initiate apoptosis through chromatin condensation and deoxyribonucleic acid fragmentation.Citation31,Citation32
Besides apoptosis, caspases have been associated with paraptosis, which is another form of programed cell death characterized by cytoplasmic vacuolation and late mitochondrial swelling.Citation33 Paraptosis has been related to phosphorylation of caspase-9 which, in turn, is associated with I/R-induced cell death.Citation23,Citation34,Citation35
Another group of signaling proteins that is strongly activated by ischemia and I/R is the SAPKs, which are closely related to the mitogen-activated protein kinases that regulate cell survival.Citation36–Citation38 SAPKs are strongly activated by ROS in the myocardium probably through activation of SAPKs kinases or inhibition of phosphoprotein phosphatases.Citation39,Citation40 It has been found that SAPK1 stimulates the mitochondrial pathway of apoptosis, which is mediated by an adenosine monophosphate-dependent activation of SAPK2.Citation41,Citation42
An additional signaling pathway that is activated by I/R is that of the protein kinase C (PKC) family. The PKC family is phospholipid-dependent kinases that are involved in the translation of signaling events ().Citation43 After ROSC, the PKCs translocate to mitochondria and other subcellular organelles and exert their ROS-mediated pro-apoptotic effects.Citation44,Citation45 Although the PKC family has been reported to signal preferentially to the SAPK1 and SAPK2 cascades and to potentiate ischemia-induced damage,Citation46 it has been suggested that translocation of PKC to mitochondria after I/R may induce cell death by stimulating the mitochondrial pathway of apoptosis.Citation47
Figure 2. Activation of programed cell death during the post-resuscitation period. PMC, protein kinase C family.
Conclusions
The post-cardiac arrest intracellular stress responses may lead to an increased threshold of oxidant-mediated injury and redox-mediated programed death of myocardial cells. Interestingly, these responses are activated during the cardiac arrest interval and are exacerbated during the CPR interval due to the massive production of ROS. More research is necessary in order to understand the molecular pathophysiology of redox-mediated programed death of myocardial cells during the post-resuscitation period.
References
- Deakin CD, Nolan JP, Soar J, Sunde K, Koster RW, Smith GB, et al. European Resuscitation Council Guidelines for Resuscitation 2010 Section 4. Adult advanced life support. Resuscitation 2101;81:1305–52.
- Chalkias A, Xanthos T. Pathophysiology and pathogenesis of post-resuscitation myocardial stunning. Heart Fail Rev 2011 May 17 [Epub ahead of print].
- Kette F, Weil MH, Gazmuri RJ, Bisera J, Rackow EC. Intramyocardial hypercarbic acidosis during cardiac arrest and resuscitation. Crit Care Med 1993;21:901–6.
- Johnson BA, Weil MH, Tang W, Noc M, McKee D, McCandless D. Mechanisms of myocardial hypercarbic acidosis during cardiac arrest. J Appl Physiol 1995;78:1579–84.
- Sharma AB, Sun J, Howard LL, Williams AG , Mallet RT. Oxidative stress reversibly inactivates myocardial enzymes during cardiac arrest. Am J Physiol Heart Circ Physiol 2007;292:H198–206.
- Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol 2003;284:H549–58.
- Niemann JT, Garner D, Lewis RJ. Tumor necrosis factor-[alpha] is associated with early postresuscitation myocardial dysfunction. Crit Care Med 2004;32:1753–58.
- Mallet RT, Sun J. Antioxidant properties of myocardial fuels. Mol Cell Biochem 2003;253:103–11.
- Buja LM. Lipid abnormalities in myocardial cell injury. Trends Cardiovasc Med 1991;1:40–5.
- Altavilla D, Saitta A, Squadrito G, Galeano M, Venuti SF, Guarini S, et al. Evidence for a role of nuclear factor-kappaB in acute hypovolemic hemorrhagic shock. Surgery 2002;131:50–8.
- Sundar SV, Li YY, Rollwagen FM, Maheshwari RK. Hemorrhagic shock induces differential gene expression and apoptosis in mouse liver. Biochem Biophys Res Commun 2005;332:688–96.
- Dos Santos P, Kowaltowski AJ, Laclau MN, Seetharaman S, Paucek P, Boudina S, et al. Mechanisms by which opening the mitochondrial ATP-sensitive K+ channel protects the ischemic heart. Am J Physiol Heart Circ Physiol 2002;283:H284–95.
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005;8:3–5.
- Sherman AJ, Davis CA, Klocke FJ, Harris KR, Srinivasan G, Yaacoub AS, et al. Blockade of nitric oxide synthesis reduces myocardial oxygen consumption in vivo. Circulation 1997;95:1328–34.
- Krismer AC, Lindner KH, Wenzel V, Rainer B, Mueller G, Lingnau W. Inhibition of nitric oxide improves coronary perfusion pressure and return of spontaneous circulation in a porcine cardiopulmonary resuscitation model. Crit Care Med 2001;29:482–6.
- Lebuffe G, Schumacker PT, Shao ZH, Anderson T, Iwase H, Vanden Hoek TL. ROS and NO trigger early preconditioning: relationship to mitochondrial KATP channel. Am J Physiol Heart Circ Physiol 2003;284:H299–308.
- Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA 2001;98:7212–7.
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000;275:25130–8.
- Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem 1998;273:18092–8.
- Solaini G, Harris DA. Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem J 2005;390:377–94.
- Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 2005;115:500–8.
- Edston E, Grontoft L, Johnsson J. TUNEL: a useful screening method in sudden cardiac death. Int J Legal Med 2002;116:22–6.
- Vanden Hoek TL, Qin Y, Wojcik K, Li CQ, Shao ZH, Anderson T, et al. Reperfusion, not simulated ischemia, initiates intrinsic apoptosis injury in chick cardiomyocytes. Am J Physiol Heart Circ Physiol 2003;284:H141–50.
- Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 1994;94:1621–8.
- Jeremias I, Kupatt C, Martin-Villalba A, Habazettl H, Schenkel J, Boekstegers P, et al. Involvement of CD95/Apo1/Fas in cell death after myocardial ischemia. Circulation 2000;102:915–20.
- Scarabelli TM, Stephanou A, Pasini E, Comini L, Raddino R, Knight RA, et al. Different signaling pathways induce apoptosis in endothelial cells and cardiac myocytes during ischemia/reperfusion injury. Circ Res 2002;90:745–8.
- Lee P, Sata M, Lefer DJ, Factor SM, Walsh K, Kitsis RN. Fas pathway is a critical mediator of cardiac myocyte death and MI during ischemia-reperfusion in vivo. Am J Physiol Heart Circ Physiol 2003;284:H456–63.
- Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res 2004;95:957–70.
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science 2004;305:626–9.
- Regula KM, Kirshenbaum LA. Apoptosis of ventricular myocytes: a means to an end. J Mol Cell Cardiol 2005;38:3–13.
- Wang X, Yang C, Chai J, Shi Y, Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 2002;298:1587–92.
- Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem 2002;277:29803–9.
- Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA 2000;97:14376–81.
- Qin Y, Vanden Hoek TL, Wojcik K, Anderson T, Li CQ, Shao ZH, et al. Caspase-dependent cytochrome c release and cell death in chick cardiomyocytes after simulated ischemia-reperfusion. Am J Physiol Heart Circ Physiol 2004;286:H2280–6.
- Lundberg KC, Szweda LI. Initiation of mitochondrial-mediated apoptosis during cardiac reperfusion. Arch Biochem Biophys 2004;432:50–7.
- Sugden PH, Clerk A. ‘Stress-responsive’ mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ Res 1998;83:345–52.
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 2001;81:807–69.
- Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, et al. MAP kinases. Chem Rev 2001;101:2449–76.
- Kyaw M, Yoshizumi M, Tsuchiya K, Kirima K, Tamaki T. Antioxidants inhibit JNK and p38 MAPK activation but not ERK 1/2 activation by angiotensin II in rat aortic smooth muscle cells. Hypertens Res 2001;24:251–61.
- Pimentel DR, Amin JK, Xiao L, Miller T, Viereck J, Oliver-Krasinski J, et al. Reactive oxygen species mediate amplitude-dependent hypertrophic, and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res 2001;89:453–60.
- Brady NR, Hamacher-Brady A, Gottlieb RA. Proapoptotic BCL-2 family members and mitochondrial dysfunction during ischaemia/reperfusion injury, a study employing cardiac HL-1 cells and GFP biosensors. Biochim Biophys Acta 2006;1757:667–78.
- Capano M, Crompton M. Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem J 2006;395:57–64.
- Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J 2003;370:361–71.
- Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c δ. Apoptosis 2003;8:19–27.
- Majumder PK, Mishra NC, Sun X, Bharti A, Kharbanda S, Saxena S, et al. Targeting PKC δ to mitochondria in the oxidative stress response. Cell Growth Differ 2001;12:465–70.
- Heidkamp MC, Bayer AL, Martin JL, Samarel AM. Differential activation of mitogen-activated protein kinase cascades and apoptosis by protein kinase C ɛ and δ in neonatal rat ventricular myocytes. Circ Res 2001;89:882–90.
- Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. Protein kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem 2004;279:47985–91.