Abstract
Objectives
During the last decade many new biological immune modulators have entered the market as new therapeutic principles. Tumor necrosis factor (TNF)-α is a pro-inflammatory cytokine known to a have a key role in the pathogenic mechanisms of various immune-mediated or inflammatory diseases. However, TNF-α also plays a key role in endothelial dysfunction and, thus, in the development and progression of atherosclerosis. What, then, is the potential therapeutic role of TNF-α inhibitors?
Methods
We analysed the current literature concerning the administration of TNF-α inhibitors and their potential benefits upon endothelial function.
Results
TNF-α inhibitors decrease the serum levels of inflammatory markers such as TNF-α itself, CRP, IL-6, and increased the tissue expression of endothelial NO synthase and the vasodilatory response to bradykinin.
Discussion
TNF-α inhibitors may change the progression of endothelial dysfunction and, thus, slow down the atherosclerotic process.
Introduction
The endothelium represents the main regulator of vascular wall homeostasis and favors a relaxed vascular tone and low levels of oxidative stress by releasing mediators such as nitric oxide (NO), prostacyclin-2, and endothelin-1, as well as by controlling local angiotensin II activity.Citation1,Citation2 In particular, NO is continuously manufactured from healthy endothelial cells through the conversion of l-arginine by the endothelial NO synthase.Citation1,Citation3 However, NO can be also produced by macrophages in response to immunological stimuli via another, inducible, NO synthase.Citation1 In this review, we describe the role of free radicals in the development of endothelial dysfunction and discuss the potential positive effects of tumor necrosis factor (TNF)-α inhibitor treatment.
Endothelial dysfunction and atherosclerosis
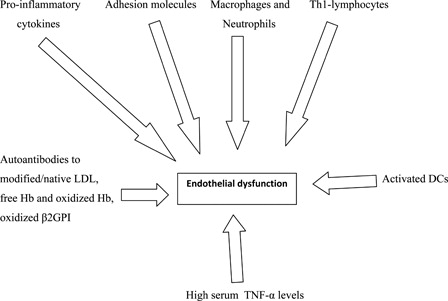
Endothelium undergoes a phenotypic modulation from the normal state to a non-adaptive state known as endothelial dysfunction in response to different noxious stimuli.Citation1 Indeed, atherosclerosis is an inflammatory condition which starts as a ‘response to injury’ that adds to traditional cardiovascular and genetic risk factors and favors the endothelial dysfunction. Early changes in endothelial function include the increase in permeability to lipoproteins and other plasma constituents, resulting in penetration of such lipids into the arterial wall and migration of monocytes and T-lymphocytes into the vessel intima.Citation1,Citation4 In particular, low density lipoproteins (LDLs) accumulate in the subendothelial space and modified or native LDL are uptaken by macrophages which become foam cells and play a key role in the development of fatty streaks.Citation1,Citation5 Pro-inflammatory cytokines such as TNF-α, interleukin (IL)-1 and IL-6, interferon-γ increase the expression of adhesion molecules including intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin and, thus, favor the proliferation of the smooth muscle cells which migrate into the lesion and lead to the thickening of the vessel wall.Citation1 The accumulation of macrophages, T-lymphocytes, smooth muscle cells, and the formation of fibrous tissue induce the enlargement and remodeling of the lesion. A fibrous cap develops over the plaque and when it becomes unstable may favor plaque rupture and thrombosis.Citation1,Citation4 The defense of a normal artery depends on innate immune responses mounted by endothelial cells and, after an inflammatory stimulus, by macrophages and other cells of the immune response. Indeed, innate immunity plays a key role in the initiation of atherosclerosis.Citation1,Citation6,Citation7 Lipid-loaded macrophages undergo apoptosis, become necrotic, and coalesce into the necrotic core of the vulnerable plaques.Citation1,Citation8 Recent evidence suggests that neutrophils also play a fundamental role in the early phase of atherosclerosis and their transmigration and degranulation in the vessel wall is determined by modified or native LDL. Finally, neutrophil inflammatory signals trigger the intimal recruitment of monocytes.Citation1,Citation9 The analysis of human atherosclerotic plaques has proven the presence of activated-T-lymphocytes expressing major histocompatibility complex class II molecules with a pro-inflammatory T-helper (Th)-1 phenotype.Citation1,Citation10 The activation of this Th-1 response represents an autoimmune mechanism in which the adaptive immune system is targeted against self-antigens expressed by atherosclerotic plaques and modified by biochemical factors as oxidative stress and hypercholesterolemia and contributes to a more aggressive progression of the atherosclerosis.Citation1,Citation11 What mechanisms can induce the autoimmune process? The answer is neither easy nor unique. However, it has been reported that microorganisms sharing sequence homology with self-molecules (molecular mimicry theory) may favor autoimmune responses,Citation1,Citation12 impairments in apoptosis, and in clearance of apoptotic bodies can render apoptotic cells as a source of autoantigens,Citation1,Citation13 CD4+ T-lymphocyte responses against native apolipoprotein B-100 may help B-lymphocytes to produce antibodies against modified or native LDLCitation1,Citation14 and, finally, defects in central tolerance favor the persistence of LDL-specific effector memory lymphocytes. A very important role is played by dendritic cells (DCs) physiologically promoting tolerization to antigens by silencing T-lymphocytes. However, danger signals may activate DCs and lead to a switch from tolerance to activation of adaptive immunity.Citation1,Citation15 Moreover, the withdrawal of the suppressive effects of regulatory T-lymphocytes may sustain inflammation and exacerbate plaque growth.Citation1 Finally, the key role of the interaction between oxidative stress and inflammation in the pathogenesis of atherosclerosis is widely accepted. It also has been proven that autoimmune responses may be directed against self-molecules altered by high affinity ligand binding or by chemical damage such as oxidative stress.Citation1,Citation16
Endothelial dysfunction, oxidative stress, and TNF-α
Arginase expressed in the endothelium serves as an endogenous competitor of NO synthase for l-arginine and, thus, plays a counteracting role in NO-mediated vasodilatory function.Citation17 It is now widely recognized that the overproduction of reactive oxygen species (ROS) and/or a deficiency of antioxidant enzyme activity may contribute to the appearance of vascular lesions.Citation1,Citation18 Indeed, ROS may catalyze several modifications to nucleic acids, lipids, and proteins favoring the generation of neo-cryptic epitopes which may behave as autoantigens.Citation1,Citation19,Citation20 Furthermore, the oxidation process is also associated with major structural LDL modifications determining the formation of new antigenic epitopes that can be presented by DCs and give rise to clonal expansion of LDL-specific T-lymphocytes, which are detectable both in serum and in human atherosclerotic plaques.Citation1,Citation16,Citation21,Citation22 Notably, autoantibodies specific for modified or native LDL have been detected in humans and have been associated with cardiovascular diseases.Citation1,Citation23 Stress-induced heat shock proteins are also considered to be autoantigens that favor the progression of atherosclerosis.Citation1,Citation24 Indeed, under stress conditions, heat shock proteins are expressed both within the cells and on the cell surface and can be released in the intercellular space, stimulating an immune response.Citation1,Citation25 Oxidative stress changes the structure and function of the β2-glycoprotein I which becomes a target antigen of a Th-1 lymphocyte response.Citation1,Citation19,Citation26 It has been demonstrated that intraplaque hemorrhages release large amounts of hemoglobin (Hb), which is oxidized in the pro-inflammatory and pro-oxidant microenvironment. Both free Hb and oxidized Hb behave as autontigens.Citation1,Citation27 TNF-α is a pro-inflammatory cytokine known to have a central role in the initial host response to infection.Citation28–Citation35 However, TNF-α up-regulates the expression of arginase in endothelial cells during ischemia/reperfusion (I/R) injury, which causes a decrease in l-arginine availability to nitric oxidase synthase and consequently leads to O2−• production.Citation17 The increased production of O2−• impairs the NO-mediated vasodilatation and favors endothelial dysfunction, which may play a key factor in the development and progression of I/R injury. These data confirm that the attenuation of NO synthesis in I/R injury may have devastating consequences.Citation17 Furthermore, hypercholesterolemia impairs the l-arginine/NO pathway, inducing activation of the angiotensin II-type 1 receptor and, thus, vasoconstriction.Citation1 Virdis et al.Citation36 studied the small resistance arteries in the visceral fat of obese patients and demonstrated a marked up-regulation of TNF-α expression mainly in the media layer of these vessels. Overall, the vascular wall represents the source of TNF-α involved in endothelial dysfunction.Citation36 More recently, a potential specific role of inflammation from perivascular adipose tissue in the pathogenesis of endothelial dysfunction has been investigated. Greenstein et al.Citation37 confirmed the increased accumulation of TNF-α in the adipose tissue, which supported the theory that in obesity the adipose tissue may damage the surrounding vessels in a paracrine manner. Notably, the vessels are not only a target but also a source of inflammation, which contributes to the pathogenesis and progression of endothelial dysfunction by activating ROS production. Furthermore, TNF-α also reduces the density of insulin receptor content at the level of the endothelial cells, worsening the insulin resistance that characterizes obese individuals and, thus, impairing the NO synthesis.Citation38 A few studies have demonstrated that women present higher serum TNF-α levels in postmenopausal period. The high levels of TNF-α associated with the low estrogen levels favor the progression of endothelial damage.Citation39,Citation40
Cells and molecules involved in endothelial dysfunction are depicted in .
Figure 1. Cells and molecules involved in endothelial dysfunction.
TNF-α inhibitors: emerging role in the prevention of endothelial dysfunction
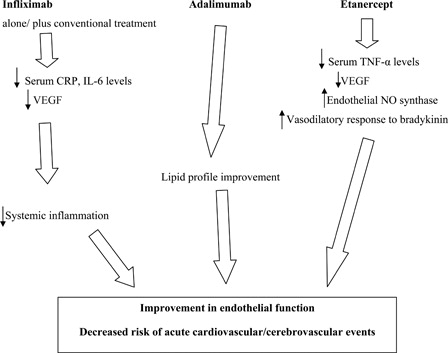
TNF-α blockers have demonstrated efficacy in large, randomized controlled clinical trials either as monotherapy or in combination with other anti-inflammatory or disease-modifying anti-rheumatic drugs in the treatment of chronic immune-mediated or inflammatory diseases.Citation41–Citation43 There are several TNF-α inhibitors available for clinical use including anti-TNF-α monoclonal antibodies (infliximab, adalimumab, golimumab, and certolizumab pegol) and a fusion protein that acts as a ‘decoy receptor’ for TNF-α (etanercept).Citation28–Citation32,Citation44 Booth et al.Citation45 confirmed that the serum levels of TNF-α may promote the progression of the endothelial dysfunction. However, C-reactive protein, TNF-α, and IL-6 increase the cardiovascular risk.Citation46–Citation49 Furthermore, TNF-α seems to favor the expression of IL-6. Therefore, on the basis of this hypothesis, they decided to treat with infliximab alone or in addition with conventional treatment for patients with anti-neutrophil cytoplasmic antibody-associated systemic vasculitis in order to address the effects of anti-inflammatory therapy upon both the systemic inflammation and endothelial dysfunction. The treatment decreased the serum levels of C-reactive protein and IL-6, but did not reduce the serum TNF-α level. Nevertheless, the current available ELISA assay measures the total TNF-α level but not those of free TNF-α which is virtually related to the disease activity. However, infliximab therapy improved both the disease activity and the endothelial function.Citation45 The forearm blood flow response to acetylcholine which resulted reduced in the patients also improved after the administration of infliximab. This finding confirms that the chronic inflammatory autoimmune diseases as systemic lupus erythematosus appear as an independent risk factor for the development of atherosclerosis and cardiovascular diseases.Citation50,Citation51 Therefore, the anti-inflammatory therapy may change both the clinical course of the autoimmune disease and the progression of the endothelial dysfunction and, thus, slow down the atherosclerotic process. Arenas et al.Citation52 evaluated the effects of etanercept administration in ovariectomized rats which presented high levels of TNF-α. Etanercept decreased the serum TNF-α level and increased the tissue expression of endothelial NO synthase and vasodilatory response to bradykinin. Dulai et al.Citation53 confirmed that TNF-α inhibitors (etanercept, infliximab, adalimumab) may have a beneficial effect on arterial stiffness in patients with rheumatoid arthritis improving, thus, accelerated atherosclerosis and consequently reducing the cardiovascular risk. Szekanecz et al.Citation54 suggested that etanercept and adalimumab may exert beneficial effects on the lipid profile improving the endothelial dysfunction. Furthermore, TNF-α inhibitors are able to reduce the expression and production of vascular endothelial growth factor (VEGF), NO, and inducible NO synthase.Citation35 VEGF is a critical mediator of inflammation both in chronic immune-mediated and allergic diseases.Citation1,Citation55,Citation56 It is known that VEGF is a pro-angiogenic factor which alters the microvascular network and, thus, correlates and may contribute to the development and progression of atherosclerosis. In summary, the administration of TNF-α inhibitors reduces the systemic inflammation in patients with chronic immune-mediated diseases, improves both the clinical course of the disease itself and the endothelial function and, thus, may decrease the risk of acute cardiovascular and/or cerebrovascular events.
Potential beneficial effects of TNF-α inhibitors are depicted in .
Figure 2. Potential beneficial effects of TNF-α inhibitors.
Conclusion
Chronic systemic inflammation that characterizes the immune-mediated or inflammatory diseases plays a key role in the development and progression of endothelial dysfunction. Free radicals such as O2−•, cytokines such as TNF-α, adhesion molecules, dyslipidemia, and innate and adaptive immune responses may favor the vascular damage. Indeed, activated Th-1 lymphocytes contribute to a more aggressive progression of atherosclerosis, which can be viewed as an autoimmune disease in which the adaptive immune system is targeted against self-antigens modified by biochemical factors such as oxidative stress. Furthermore, TNF-α up-regulates the expression of arginase in endothelial cells and leads to O2−• production that impairs the NO-mediated vasodilatation and favors endothelial dysfunction. Notably, the vascular wall represents the source of TNF-α involved in endothelial dysfunction as demonstrated in obese individuals. TNF-α inhibitors by decreasing systemic inflammation and production of VEGF seem to have a beneficial effect upon the progression of atherosclerosis, reducing the risk of acute cardiovascular and/or cerebrovascular events.
References
- Murdaca G, Colombo BM, Cagnati P, Gulli R, Spanò F, Puppo F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012;224(2):309–17.
- Gonzalez MA, Selwyn AP. Endothelial function, inflammation, and prognosis in cardiovascular disease. Am J Med 2003;115 (Suppl 8A):99S–106.
- Napoli C, Ignarro LJ. Nitric oxide and atherosclerosis. Nitric Oxide 2001;5(2):88–97.
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 2011;473(7347):317–25.
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 2007;116(16):1832–44.
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol 2011;12(3):204–12.
- Lundberg AM, Hansson GK. Innate immune signals in atherosclerosis. Clin Immunol 2010;134:5–24.
- Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 2010;10:36–46.
- Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 2011;17(11):1410–22.
- Jonasson L, Holm J, Skalli O. Expression of class II transplantation antigen on vascular smooth muscle cells in human atherosclerosis. J Clin Invest 1985;76(1):125–31.
- Nilsson J, Wigren M, Shah PK. Regulatory T cells and the control of modified lipoprotein autoimmunity-driven atherosclerosis. Trends Cardiovasc Med 2009;19(8):272–6.
- Blank M, Shoenfeld Y. Beta-2-glycoprotein-I, infections, antiphospholipid syndrome and therapeutic considerations. Clin Immunol 2004;112(2):190–9.
- Hall JC, Casciola-Rosen L, Rosen A. Altered structure of autoantigens during apoptosis. Rheum Dis Clin North Am 2004;30(3):455–71.
- Hermansson A, Ketelhuth DF, Strodthoff D, Wurm M, Hansson EM, Nicoletti A, et al. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J Exp Med 2010;207(5):1081–93.
- Niessner A, Weyand CM. Dendritic cells in atherosclerotic disease. Clin Immunol 2010;134:25–32.
- Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, Hansson GK. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci USA 1995;92(9):3893–7.
- Gao X, Xu X, Belmadani S, Park Y, Tang Z, Feldman AM, et al. TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol 2007;27(6):1269–75.
- Puddu P, Puddu GM, Cravero E, De Pascalis S, Muscari A. The emerging role of cardiovascular risk factor-induced mitochondrial dysfunction in atherogenesis. J Biomed Sci 2009;16:112.
- Buttari B, Profumo E, Mattei V, Siracusano A, Ortona E, Margutti P, et al. Oxidized b-glycoprotein I induces human dendritic cell maturation and promotes a T helper type 1 response. Blood 2005;106(12):3880–7.
- Arvieux J, Regnault V, Hachulla E, Darnige L, Berthou F, Youinou P. Oxidation of b2-glycoprotein I (b2-GPI) by the hydroxil radical alters phospholipid binding and modulates recognition by b2-GPI autoantibodies. Thromb Haemost 2001;86(4):1070–6.
- Nilsson J, Hansson GK. Autoimmunity in atherosclerosis: a protective response losing control? J Intern Med 2008;263(5):464–78.
- Frostegard J, Wu R, Giscombe R, Holm G, Lefvert AK, Nilsson J. Induction of T-cell activation by oxidized low density lipoprotein. Arterioscler Thromb 1992;12(4):461–7.
- Nilsson J, Kovanen PT. Will autoantibodies help to determine severity and progression of atherosclerosis? Curr Opin Lipidol 2004;15(5):499–503.
- Wick G, Knoflach M, Xu Q. Autoimmune and inflammatory mechanisms in atherosclerosis. Annu Rev Immunol 2004;22:361–403.
- Park HK, Park EC, Bae SW, Park MY, Kim SW, Yoo HS, et al. Expression of heat shock protein 27 in human atherosclerotic plaques and increased plasma level of heat shock protein 27 in patients with acute coronary syndrome. Circulation 2006;114(9):886–93.
- Profumo E, Buttari B, Alessandri C, Conti F, Capoano R, Valesini G, et al. Beta2-glycoprotein I is a target of t cell reactivity in patients with advanced carotid atherosclerotic plaques. Int J Immunopathol Pharmacol 2010;23(1):73–80.
- Buttari B, Profumo E, Petrone L, Pietraforte D, Siracusano A, Margutti P, et al. Free hemoglobin: a dangerous signal for the immune system in patients with carotid atherosclerosis? Ann N Y Acad Sci 2007;1107:42–50.
- Murdaca G, Colombo BM, Cagnati P, Gulli R, Spanò F, Puppo F. Update upon efficacy and safety of TNF-α inhibitors. Expert Opin Drug Saf 2012;11(1):1–5.
- Murdaca G, Colombo BM, Puppo F. Emerging biological drugs: a new therapeutic approach for systemic lupus erythematosus. An update upon efficacy and adverse events. Autoimmun Rev 2011;11(1):56–60.
- Murdaca G, Colombo BM, Puppo F. Adalimumab for the treatment of immune-mediated diseases: an update on old and recent indications. Drugs Today (Barc) 2011;47(4):277–88.
- Murdaca G, Colombo BM, Puppo F. Anti-TNF-alpha inhibitors: a new therapeutic approach for inflammatory immune-mediated diseases: an update upon efficacy and adverse events. Int J Immunopathol Pharmacol 2009;22(3):557–65.
- Murdaca G, Colombo BM, Barabino G, Caiti M, Cagnati P, Puppo F. Anti-tumor necrosis factor-α treatment with infliximab for disseminated granuloma annulare. Am J Clin Dermatol 2010;11(6):437–9.
- Murdaca G, Colombo BM, Contini P, Puppo F. Determination of lymphotoxin-alpha levels in patients with psoriatic arthritis undergoing etanercept treatment. J Interferon Cytokine Res 2012;32(6):277–9.
- Murdaca G, Spanò F, Puppo F. Selective TNF-α inhibitor-induced injection site reactions. Expert Opin Drug Saf 2013;12(2):187–93.
- Murdaca G, Spanò F, Miglino M, Puppo F. Effects of TNF-α inhibitors upon the mechanisms of action of VEGF. Immunotherapy 2013;5(2):113–5.
- Virdis A, Santini F, Colucci R, Duranti E, Salvetti G, Rugani I, et al. Vascular generation of tumor necrosis factor-α reduces nitric oxide availability in small arteries from visceral fat of obese patients. J Am Coll Cardiol 2011;58(3):238–47.
- Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 2009;119:1661–70.
- Aljada A, Ghanim H, Assian E, Dandona P. Tumor necrosis factor-alpha inhibits insulin-induced increase in endothelial nitric oxide synthase and reduces insulin receptor content and phosphorylation in human aortic endothelial cells. Metabolism 2002;51:487–91.
- Kamada M, Irahara M, Maegawa M, Ohmoto Y, Takeji T, Yasui T, et al. Postmenopausal changes in serum cytokine levels and hormone replacement therapy. Am J Obstet Gynecol 2001;184:309–14.
- Sites CK, Toth MJ, Cushman M, L'Hommedieu GD, Tchernof A, Tracy RP, et al. Menopause-related differences in inflammation markers and their relationship to body fat distribution and insulin-stimulated glucose disposal. Fertil Steril 2002;77(1):128–35.
- Taylor PC. Antibody therapy for rheumatoid arthritis. Curr Opin Pharmacol 2003;3:323–8.
- Chang J, Girgis L. Clinical use of anti-TNF-alpha biological agents – a guide for GPs. Aust Fam Physician 2007;36:1035–8.
- Puppo F, Murdaca G, Ghio M, Indiveri F. Emerging biologic drugs for the treatment of rheumatoid arthritis. Autoimmun Rev 2005;4(8):537–41.
- Hochberg MC, Lebwohl MG, Plevy SE, Hobbs KF, Yocum DE. The benefit/risk profile of TNF-blocking agents: findings of a consensus panel. Semin Arthritis Rheum 2005;34:819–36.
- Booth AD, Jayne DR, Kharbanda RK, McEniery CM, Mackenzie IS, Brown J, et al. Infliximab improves endothelial dysfunction in systemic vasculitis: a model of vascular inflammation. Circulation 2004;109(14):1718–23.
- Morrow DA, Rifai N, Antman EM, Weiner DL, McCabe CH, Cannon CP, et al. C-reactive protein is a potent predictor of mortality independently of and in combination with troponin T in acute coronary syndromes: a TIMI 11A substudy. Thrombolysis in myocardial infarction. J Am Coll Cardiol 1998;31(7):1460–5.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997;336:973–9.
- Ridker PM, Rifai N, Pfeffer M, Sacks F, Lepage S, Braunwald E. Elevation of tumor necrosis factor-alpha and increased risk of recurrent coronary events after myocardial infarction. Circulation 2000;101:2149–53.
- Biasucci LM, Vitelli A, Liuzzo G, Altamura S, Caligiuri G, Monaco C, et al. Elevated levels of interleukin-6 in unstable angina. Circulation 1996;94:874–7.
- Colombo BM, Cacciapaglia F, Puntoni M, Murdaca G, Rossi E, Rodriguez G, et al. Traditional and non traditional risk factors in accelerated atherosclerosis in systemic lupus erythematosus: role of vascular endothelial growth factor (VEGATS Study). Autoimmun Rev 2009;8(4):309–15.
- Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, du Berger R, et al. Traditional risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001;44:2331–7.
- Arenas IA, Armstrong SJ, Xu Y, Davidge ST. Chronic tumor necrosis factor-alpha inhibition enhances NO modulation of vascular function in estrogen-deficient rats. Hypertension 2005;46(1):76–81.
- Dulai R, Perry M, Twycross-Lewis R, Morrissey D, Atzeni F, Greenwald S. The effect of tumor necrosis factor-α antagonists on arterial stiffness in rheumatoid arthritis: a literature review. Semin Arthritis Rheum 2012;42(1):1–8.
- Szekanecz Z, Kerekes G, Soltész P. Vascular effects of biologic agents in RA and spondyloarthropathies. Nat Rev Rheumatol 2009;5(12):677–84.
- Ciprandi G, Murdaca G, Colombo BM, De Amici M, Marseglia GL. Serum vascular endothelial growth factor in allergic rhinitis and systemic lupus erythematosus. Hum Immunol 2008;69(8):510–2.
- Ciprandi G, Colombo BM, Murdaca G, De Amici M. Serum vascular endothelial growth factor and sublingual immunotherapy. Allergy 2008;63(7):945–6.