Abstract
Nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) are the most common underlying causes of chronic liver injury. They are associated with a wide spectrum of hepatic disorders including basic steatosis, steatohepatitis, and cirrhosis. The molecular and cellular mechanisms underlying hepatic injury in NAFLD and NASH are still unknown. This review describes the roles of oxidative stress and inflammatory responses in the pathogenesis of NAFLD and its progression to NASH.
Introduction
Nonalcoholic fatty liver diseases (NAFLDs) are a group of chronic liver disorders which have garnered attention in recent years as emerging health problems worldwide. Up to 35% of adults in the United States are believed to be affected. The clinical spectrum of NAFLDs ranges from simple fatty change to nonalcoholic steatohepatitis (NASH), which is characterized by cytolytic changes in hepatocytes (i.e. ballooning degeneration, Mallory bodies, and lobular inflammation). Although NAFLD is mostly benign, 20–30% of the patients develop NASH which may eventually progress to hepatic cirrhosis.Citation1,Citation2
While the reason why some patients with NAFLD show signs of chronic liver injury, while others do not remains elusive, a ‘two-hit’ theory has been postulated to help explain the mechanisms behind the development of advanced NAFLD. The ‘first hit’ is fat accumulation which is believed to sensitize the liver, triggering an inflammatory process that results in steatohepatitis and fibrosis.Citation3 Although the exact nature of the ‘second hit’ has yet to be fully described, extensive research has identified several possible mechanisms, including oxidative stress (OS)-induced inflammation with lipid peroxidation, cytokine activation as well as excess production of reactive oxygen and nitrogen species (ROS/RNS).Citation4 The association between insulin resistance (IR) and hepatocellular fat accumulation has also been well documented.Citation5
The role of OS in NAFLD/NASH remains poorly understood, and it is widely believed that elucidation of the pathophysiological mechanisms involved may help to develop noninvasive diagnostic and therapeutic options for this condition. This article offers an updated review of current researches on the role of OS and anti-oxidant mechanisms in the development of NAFLD/NASH.
The synthesis and cellular sources of ROS
Molecular oxygen, which readily accepts electrons, is the main source of radicals in biological systems, the most important of which being the hydroxyl radical (•OH), nitric oxide radical (NO•) and the superoxide anion (O2•−). These unstable and reactive radicals are natural by products of intracellular metabolism and from exogenous substances, which have the ability to react with biological compounds including proteins, free fatty acids (FFA), and DNA.Citation6 While the main endogenous intracellular sources of ROS are mitochondria, endoplasmic reticulum (ER), and peroxisomes, superoxide anion radicals (O2•−) are produced as a result of enzymatic activity, such as with xanthine oxidase (XO) and cytochrome P450 metabolism.Citation7,Citation8 Normally, there is a fine balance between pro-oxidant and antioxidant mechanisms, and OS has long been recognized as a key mechanism responsible for liver damage and disease progression in NAFLD, is believed to occur due to an imbalance in favor of prooxidation.Citation9 Accumulating evidence suggests that mitochondrial dysfunction plays a significant role in steatosis and steatohepatitis. Mitochondrial dysfunction results in ROS overproduction, and the ensuing increase in lipid peroxidation and protein oxidation has a detrimental effect on fat homeostasis in the liver. Mitochondria remain the main source of ROS in hepatocytes, although other subcellular organelles have also been shown to participate in this process.Citation9,Citation10 In fact, peroxisomes are able to oxidize long-chain FFA more rapidly than mitochondria, thus increasing a cell's capacity to metabolize FFA. However, H2O2 which is an end-product of peroxisomal β-oxidation is easily converted into the highly reactive OH radical.Citation10 Chronic ER stress may also contribute to OS, by promoting toxic accumulation of ROS which trigger other signaling pathway within the cell. The relationship between ER stress and OS works both ways since ROS generated through inflammation or damage to organelles (e.g. mitochondria) may also accelerate ER dysfunction.Citation11,Citation12 The toxic effects of long-chain fatty acids are circumvented by lipo-oxygenation via microsomal cytochromes, CYP2E1 and CYP4A, with subsequent release of ROS. Microsomal and peroxisomal oxidation normally play a minor role in the metabolism of fatty acid, but may become more involved when low CYP2E1 levels result in accumulation of long-chain fatty acids.Citation13,Citation14 Emery et al.Citation15 have documented that CYP2E1 levels increased in morbidly obese patients with NAFLD.
ROS may also interact with polyunsaturated fatty acids (PUFAs). The resultant induction of lipid peroxidation within the cell leads to the formation of 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) (), which have longer half-lives than ROS, but can freely diffuse into the extracellular space to affect distant cells, therefore amplifying the effects of OS. It would seem that all endogenous intracellular sources of ROS (mitochondria, XO, ER, CYP2E1, and peroxisomes) contribute to disease progression in NAFLD.
Table 1. Potential noninvasive diagnostic biomarkers of hepatic oxidative stress in NAFLD/NASH
Decreased antioxidant defense and increased lipid peroxidation in liver cells
Oxidative stress occurs as a result of either excessive production of ROS within the hepatocyte or reduced antioxidant defences. The major antioxidant enzymes, copper/zinc superoxide dismutase (Cu/Zn SOD) and manganese-superoxide dismutase (Mn-SOD), which are mainly present in the cytoplasm and mitochondria, promote the reduction of O2•− to H2O2. Another antioxidant enzyme, glutathione peroxidase (GPx), facilitates the subsequent conversion of H2O2 into H2O.Citation6,Citation16 A breakdown in antioxidant defenses has been proven to play a significant role in OS associated with NASH, as evidenced by decreased hepatic glutathione (GSH) and diminished SOD, GPx, catalase, and glutathione transferase activities in correlation with disease severity.Citation8,Citation17 Yesilova et al. observed an increase in serum oxidative markers (e.g. MDA) which was paralleled by a decrease in the activity of antioxidants (coenzyme Q10, Cu/Zn SOD, and catalase activity) in patients with NAFLD. Investigators also reported on a significant correlation between severity of liver disease and IR.Citation18 Low levels of mitochondrial GSH have also been reported in a murine model of NASH, and it has been suggested that increased levels of cholesterol within the inner mitochondrial membrane may have contributed to this reduction.Citation8
Thioredoxin (Trx) plays an essential role in cell function as it limits OS directly via its antioxidant properties and also indirectly by proteins interaction with key signal transduction molecules. Sumida et al. reported on higher serum levels of Trx in patients with NASH compared to those with simple steatosis or healthy volunteers.Citation19 A similar result was also reported in a comparison between patients with NASH and healthy controls.Citation20
NAFLD is characterized by fat accumulation in hepatocytes, which provides a potential substrate for lipid peroxidation and ROS toxicity. Ensuing excessive ROS production enhances lipid peroxidation which subsequently leads to the formation of other reactive metabolites in the liver, such as 4-HNE and MDA. IR is another contributing factor, leading to increased production of ROS in the liver, higher levels of hepatic lipid peroxidation, and decreased antioxidant capacity in the plasmaCitation21,Citation22 ().
Figure 1. Basic mechanistic model associated with oxidative stress in the pathogenesis of NAFLD and progression to NASH.
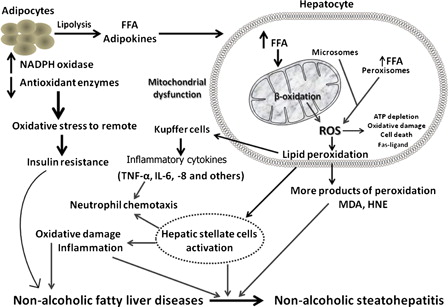
Mitochondrial dysfunction in NAFLD
Mitochondrial dysfunction and OS play a central role in the pathophysiology of NAFLD/NASH, regardless of initial cause (e.g. drug-induced or IR-related), although the underlying mechanisms remain unclear. They have been shown to participate at various levels in NASH pathogenesis, by impairing fat metabolism and consequently increasing OS and cytokine production, which trigger cell death and promote inflammation.Citation8,Citation9 The formation of ROS and RNS, GSH depletion and protein oxidation are major events associated with mitochondrial dysfunction, which represent critical initiating events for the development of NAFLD.Citation23 The loss of mitochondrial function leads to secondary inhibition of the β-oxidation of lipids, further increasing the steatosis and starting a vicious circle. This is further complicated by the presence of obesity and IR, which in themselves are associated with an imbalance between uptake/synthesis and oxidation/export of fatty acids and accumulation of triglycerides in hepatocytes.Citation9,Citation24 ROS generation in a lipid-rich environment in turn induces lipid peroxidation and which may lead to the release of highly reactive aldehydic derivatives (e.g. MDA) that have diverse detrimental effects on hepatocytes and other liver cells. ROS and other products of lipid peroxidation further impair the respiratory chain in hepatocytes which is also an important subcellular source of ROS. Impaired oxidative phosphorylation resulting in reduced hepatic ATP synthesis and increased ROS production has been reported in patients with NASH.Citation9,Citation25 Uncoupling protein 2 (UCP-2) is an inner mitochondrial protein that becomes abundant in hepatocytes of fatty liver, and its expression in physiological circumstances is mainly limited to Kupffer cells. Increased expression of UCP-2 in the livers of patients with NAFLD has been shown to reflect impaired oxidative phosphorylation.Citation8
Alterations in mitochondrial morphology, impaired mitochondrial bioenergetics, increased mitochondrial lipid peroxides, and decreased ATP content have also been described in a variety of models of fatty liver disease.Citation26,Citation27 The peroxisome proliferator activated receptor-α (PPAR-α) regulates the expression of mitochondrial and peroxisomal β-oxidation enzymes involved in the metabolism of lipids. PPAR-α is activated by long-chain FFAs, and its activation also increases expression of UCP-2 mRNA. Increased expression of hepatic PPAR-α mRNA has also been demonstrated in different genetic models of obesity and massive steatosis.Citation8,Citation28
Under normal physiological conditions, fatty acids are deposited predominantly via mitochondrial oxidative pathways. Significant increase in mitochondrial oxidation is believed to be the main source of ROS in NAFLDCitation6 (). The resultant increase in mitochondrial ROS augments lipid peroxidation to release more reactive aldehydes, further damaging mitochondrial DNA (mtDNA) and respiratory chain polypeptides.Citation29 In summary, mitochondrial dysfunction not only impairs fat homeostasis in the liver but also leads to an overproduction of ROS which is considered to be an important factor in producing lethal hepatocyte injury associated with NAFLD.Citation8,Citation30
Effects of inflammation and oxidative stress in NASH
Recent studies have suggested several possibilities, including inflammation caused by OS associated with lipid peroxidation, cytokine activation, NO and endogenous toxins of fructose metabolites.Citation3,Citation4 Mitochondrial dysfunction not only facilitated the production of ROS, but also contributes to the progression of NAFLD by inducing hepatic inflammatory cytokines. It has been postulated that obesity, IR, and adipokine/cytokine networks induce both liver fat accumulation and NASH development.Citation31 ROS along with products of lipid peroxidation result in increased release of several cytokines (tumor necrosis factor-alpha (TNF-α), Fas ligand) which playing a key role in cell death, inflammation, and fibrosisCitation8,Citation9 (). Besides the membrane damage caused by lipid peroxidation, protein damage has also been shown to develop as a consequence of OS. The aldehyde end products of lipid peroxidation are well-documented proinflammatory mediators which activate stellate cells, leading to increased collagen synthesis and subsequent development of liver fibrosis.Citation32 Although higher levels of lipid peroxidation activity have been demonstrated in the livers of patients with NASH compared to those with simple steatosis, results of studies investigating the association between systemic levels of lipid peroxidation products and hepatic necroinflammatory activity have been inconsistent.Citation29
Lipid peroxidation, release of inflammatory cytokines, and cell death are the consequences of ROS-mediated mechanisms. Biologically active lipid peroxidation products and cytokines act together by inducing hepatic inflammation leading to the development of diverse hepatic lesions associated with NASH.Citation8 The inflammatory response is induced as a result of upregulation of pro-inflammatory cytokines including TNF-α, interleukin (IL) 1, and IL-6,Citation33,Citation34 which play an important role in directing polymorpho- and mono-nuclear leukocytes into inflamed tissues. Research on serum levels of TNF-α and IL-6 in patients with NASH has so far produced inconsistent results.Citation35 A positive correlation has been reported between hepatic IL-6 expression, stage of fibrosis, and degree of systemic IR.Citation36,Citation37 Moreover, activation of Kupffer cells and expression of death receptor Fas-ligandCitation12 has been shown to be associated with increased necrosis via activation of caspases and Fas-ligand mediated cell death.Citation14,Citation38
The effects of TNF-α in NASH are enhanced through an abnormal cytokine profile, and increased expression of the TNF-α-receptor in the liver.Citation39 This contributes to additional lipid peroxidation of mitochondrial membranes, hence worsening their function and further inducing OS.Citation38 Adipose tissue shows prominent deregulation of genes related to inflammation in patients with NASH. Induction of NADPH oxidase by TNF-α also leads to inflammation through TNF receptor-1 expression and nuclear factor kappa B (NF-κB) activation.Citation14,Citation40
The association of elevated serum iron values and increased hepatic tissue ferritin deposition with hepatic inflammation and IR in patients with NASH has been well established.Citation34,Citation41 On the other hand, ROS and lipid peroxidation cause direct damage to hepatocytes by affecting membranes, proteins, and DNA.Citation42 Ensuing damage to nuclear and mtDNA results in necro-inflammation, particularly in the nuclei/cytoplasm of hepatocytes and sinusoidal cells.Citation42,Citation43 ROS also trigger NF-κB activation, which in turn induces triggers TNF-α synthesis and consequently mtDNA damage.Citation44 Increased levels of 8-hydroxy-2′deoxyguanosine (8-OHdG), which is a modified DNA base product generated by ROS, are indicative of due to nuclear and mtDNA damage, and have been demonstrated to correlate with grade of inflammation.Citation42
Hepatic inflammation and fibrogenic progression are pivotal features in the pathogenesis of steatohepatitis. Although hepatocyte damage and OS are believed to be the initial triggers of inflammation, additional factors such as mitochondrial dysfunction and ER stress have also been implicated as contributory factors in the progression of NAFLD to NASH, by promoting the generation of signals and mediators of inflammation.
Markers of iron metabolism in NAFLD/NASH
In recent years, a strong link has been demonstrated between iron overload and several manifestations of the metabolic syndrome including NAFLD. It has been argued that increased ferritin levels observed in the majority of patients with NASH is due to the underlying necro-inflammatory condition which facilitates the release of tissue iron and ferritin into the blood.Citation25,Citation45 Mendler et al.Citation46 were the first to suggest the presence of a possible link between hepatic iron-overload and IR. The association between NASH and IR has recently been described as a ‘new iron overload syndrome’ characterized by hyperferritinemia. The dysmetabolic iron overload syndrome has now been established as a frequent finding in the general population, occurring in about one-third of patients with NAFLD and the metabolic syndrome. Altered regulation of iron transport associated with steatosis, IR, and subclinical inflammation, often in the presence of predisposing genetic factors are considered the main contributors to iron overload.Citation47 Although iron-associated OS is believed to play at least a partial role in the pathogenesis in NAFLD/NASH, the exact mechanisms behind deposition of hepatic iron remain unknown. One of the clinical features of NASH is the buildup of iron in the liver accompanied by increased level of serum ferritin, which is highly suggestive of a central role of iron in the disease process.Citation34,Citation48 It is well known that iron generates highly reactive hydroxyl radicals through the Fenton reaction, and the resultant ROS may contribute to liver damage.Citation6 While ROS induce peroxidation of PUFAs and nucleic acids,Citation49 lipophilic antioxidants, such as vitamin E, have been shown to decrease hepatocellular damage in NASH.Citation47. A role of iron overload in oxidative DNA damage has also been suggested. Significant increases in hepatic 8-OHdG generated by OH radicals have been reported in patients with NASH, particularly in correlation with iron overload, IR, and severity of hepatic steatosis.Citation47,Citation50
Diagnostic biomarkers and therapeutic targets
The value of research so far has been limited by lack of reproducibility or failure to accurately distinguish basic steatosis from more advanced situations. Investigators have stressed on a need for the discovery of new biomarkers in an effort to support a diagnosis of NAFLD.Citation51 To date, there is no routine biomarker for NAFLD and liver biopsy remains the gold standard for diagnosis. Nevertheless, there is a growing consensus that NAFLD could be diagnosed accurately without the need for liver biopsy, using a combination of clinical history, laboratory tests, and ultrasound.Citation52
Previous studies have demonstrated elevated levels of MDA and 4-HNE in up to 90% of NASH patients compared to those with steatosis, a result indicative of increased OS. Levels of 4-HNE, which is mainly found in hepatocyte cytoplasm and sinusoidal cells, have been shown to correlate with grade of inflammation. Other noninvasive markers of OS that have been proposed include isoprostanes and 3-nitrotyrosine (3-NT), the increased activities of which are believed to be indicative of hepatic lipid peroxidation. Liver 3-NT reactivity, a marker of RNS-induced stress, has been shown to increase significantly in the presence of steatosis.Citation29 F2-isoprostanes are produced by nonenzymatic peroxidation of arachidonic acid, and increased plasma and urine levels are widely regarded as valuable biomarkers of lipid peroxidation.Citation2 Significant elevations in F2-isoprostanes have been reported in patients with NASHCitation53 as well as in rat NAFLD models, findings which support a role of OS in the steatotic liver.
Elevated 8-OHdG content in hepatic mtDNA has been demonstrated in patients with NAFLDCitation42. Furthermore, higher levels of 8-OHdG expression in the liver as well as urinary excretion has been reported in patients with NASH compared to healthy controls.Citation54
Multiple interrelated factors such as IR, OS, inflammation, and genetic predisposition, all contribute to the development of NAFLD, each of which is potential therapeutic target. Lower levels of serum antioxidants are present in patients with NASH, and depletion of antioxidants via lipid peroxidation and ROS renders the liver more susceptible to oxidative damage. When administered, the lipophilic antioxidant vitamin E acts to counteract this decrease in levels of antioxidant enzymes, thus slowing progression of NAFLD to NASH. A beneficial effect of vitamin E was demonstrated in steatohepatitis mice fed with a methionine–choline-deficient diet, evidenced by a decrease in liver enzymes as well as a reduction in histological steatosis and necroinflammation scores. The naturally occurring thiol antioxidant, alpha lipoic acid, has been shown to have a hepatoprotective effect associated with reduced expression of CYP2E1 and decreased ER stress, while also reducing the activity of mitogen-activated protein kinases and NF-κB.Citation55
Conclusion
The undeniable role of OS in the pathogenesis of NAFLD has been evidenced by the detection of significant increases in markers of oxidative damage of lipids (MDA, 4-HNE), proteins (3-NT content), and DNA (8-OH-dG), paralleled by a reduction in antioxidant mechanisms, including a decrease in levels of coenzyme Q10 and GSH levels as well as a reduction in the activity of SOD, GPx, and catalase. Other supporting findings include an increase in Trx and the induction of iNOS and CYP2E1.Citation18,Citation19,Citation42 These changes are manifested by a significant increase in serum levels of MDA and 4-HNE levels, as has been demonstrated in patients with steatohepatitis compared to those with simple steatosis.Citation56 The importance of the above contention is highlighted by the lack of a drug therapy that is effective for NAFLD at present,Citation57 suggesting potentially attractive therapeutic targets to improve insulin sensitivity in obese NAFLD patients. Better understanding of the role of OS and inflammatory response in the pathogenesis of NAFLD and NASH would help in the development new management strategies.
References
- Lazo M, Clark JM. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Semin Liver Dis 2008;28(4):339–50.
- Reid AE. Nonalcoholic steatohepatitis. Gastroenterology 2001;121(3):710–23.
- Day CP, James OFW. Steatohepatitis: a tale of two ‘hits’?. Gastroenterology 1998;114(4):842–5.
- Nomura K, Yamanouchi T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J Nutr Biochem 2012;23(3):203–8.
- Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107(5):450–5.
- Gutteridge JM, Halliwell B. Free radicals and antioxidants in the year 2000. A historical look to the future. Ann N Y Acad Sci 2000;899:136–47.
- Robertson G, Leclercq I, Farrell GC. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am J Physiol Gastrointest Liver Physiol 2001;281(5):G1135–9.
- Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 2012;52(1):59–69.
- Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion 2006;6(1):1–28.
- Koek GH, Liedorp PR, Bast A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin Chim Acta 2011;412(15–16):1297–305.
- Bonekamp NA, Völkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: struggling for balance. Biofactors 2009;35(4):346–55.
- Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol 2006;38(3):317–32.
- Chalasani N, Gorski JC, Asghar MS, Asghar A, Foresman B, Hall SD, et al. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 2003;37(3):544–50.
- Malaguarnera M, Di Rosa M, Nicoletti F, Malaguarnera L. Molecular mechanisms involved in NAFLD progression. J Mol Med (Berl) 2009;87(7):679–95.
- Emery MG, Fisher JM, Chien JY, Kharasch ED, Dellinger EP, Kowdley KV, et al. CYP2E1 activity before and after weight loss in morbidly obese subjects with nonalcoholic fatty liver disease. Hepatology 2003;38(2):428–435.
- Akyol O, Herken H, Uz E, Fadillioğlu E, Unal S, Söğüt S, et al. The indices of endogenous oxidative and antioxidative processes in plasma from schizophrenic patients. The possible role of oxidant/antioxidant imbalance. Prog Neuropsychopharmacol Biol Psychiatry 2002;26(5):995–1005.
- Hardwick RN, Fisher CD, Canet MJ, Lake AD, Cherrington NJ. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos 2010;38(12):2293–301.
- Yesilova Z, Yaman H, Oktenli C, Ozcan A, Uygun A, Cakir E, et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am J Gastroenterol 2005;100(4):850–5.
- Sumida Y, Nakashima T, Yoh T, Furutani M, Hirohama A, Kakisaka Y, et al. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. Hepatol 2003;38(1):32–8.
- Nakashima T, Sumida Y, Furutani M, Hirohama A, Okita M, Mitsuyoshi H, et al. Elevation of serum thioredoxin levels in patients with nonalcoholic steatohepatitis. Hepatol Res 2005;33(2):135–7.
- Videla LA, Rodrigo R, Araya J, Poniachik J. Insulin resistance and oxidative stress interdependency in non-alcoholic fatty liver disease. Trends Mol Med 2006;12(12):555–8.
- Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, et al. Increase in long-chain polyunsaturated fatty acid n− 6/n−3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004;106(6):635–43.
- Grattagliano I, de Bari O, Bernardo TC, Oliveira PJ, Wang DQ, Portincasa P. Role of mitochondria in nonalcoholic fatty liver disease–from origin to propagation. Clin Biochem 2012;45(9):610–8.
- Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol 2005;42(6):928–40.
- Pérez-Carreras M, Del Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38(4):999–1007.
- Chavin KD, Yang S, Lin HZ, Chatham J, Chacko VP, Hoek JB, et al. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J Biol Chem 1999;274(9):5692–700.
- Teodoro JS, Rolo AP, Duarte FV, Simões AM, Palmeira CM. Differential alterations in mitochondrial function induced by a choline-deficient diet: understanding fatty liver disease progression. Mitochondrion 2008;8(5–6):367–76.
- Nakatani T, Tsuboyama-Kasaoka N, Takahashi M, Miura S, Ezaki O. Mechanism for peroxisome proliferator-activated receptor-alpha activator-induced up-regulation of UCP2 mRNA in rodent hepatocytes. J Biol Chem 2002;277(11):9562–9.
- Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001;120(5):1183–92.
- Oliveira CP, da Costa Gayotto LC, Tatai C, Della Bina BI, Janiszewski M, Lima ES, et al. Oxidative stress in the pathogenesis of nonalcoholic fatty liver disease, in rats fed with a choline-deficient diet. J Cell Mol Med 2002;6(3):399–406.
- Petta S, Muratore C, Craxì A. Non-alcoholic fatty liver disease pathogenesis: the present and the future. Dig Liver Dis 2009;41(9):615–25.
- Lee KS, Buck M, Houglum K, Chojkier M. Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest 1995;96(5):2461–8.
- Armutcu F, Coskun O, Gürel A, Kanter M, Can M, Ucar F, et al. Thymosin alpha 1 attenuates lipid peroxidation and improves fructose-induced steatohepatitis in rats. Clin Biochem 2005;38(6):540–7.
- Uysal S, Armutcu F, Aydogan T, Akin K, Ikizek M, Yigitoglu MR. Some inflammatory cytokine levels, iron metabolism and oxidan stress markers in subjects with nonalcoholic steatohepatitis. Clin Biochem 2011;44(17–18):1375–9.
- Rabelo F, Oliveira CP, Faintuch J, Mazo DF, Lima VM, Stefano JT, et al. Pro- and anti-inflammatory cytokines in steatosis and steatohepatitis. Obes Surg 2010;20(7):906–12.
- Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol 2008;103(6):1372–9.
- Estep JM, Baranova A, Hossain N, Elariny H, Ankrah K, Afendy A, et al. Expression of cytokine signaling genes in morbidly obese patients with non-alcoholic steatohepatitis and hepatic fibrosis. Obes Surg 2009;19(5):617–24.
- Syn WK, Choi SS, Diehl AM. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clin Liver Dis 2009;13(4):565–80.
- Diehl AM. Nonalcoholic steatosis and steatohepatitis IV. Nonalcoholic fatty liver disease abnormalities in macrophage function and cytokines. Am J Physiol Gastrointest Liver Physiol 2002;282(1):G1–5.
- Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003;125(2):437–43.
- Fujita N, Takei Y. Iron Overload in Nonalcoholic Steatohepatitis. Adv Clin Chem 2011;55:105–32.
- Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol 2002;37(1):56–62.
- Nomoto K, Tsuneyama K, Takahashi H, Murai Y, Takano Y. Cytoplasmic fine granular expression of 8-hydroxydeoxyguanosine reflects early mitochondrial oxidative DNA damage in nonalcoholic fatty liver disease. Appl Immunohistochem Mol Morphol 2008;16(1):71–5.
- Nagakawa Y, Williams GM, Zheng Q, Tsuchida A, Aoki T, Montgomery RA, et al. Oxidative mitochondrial DNA damage and deletion in hepatocytes of rejecting liver allografts in rats: role of TNF-alpha. Hepatology 2005;42(1):208–15.
- Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999;30(6):1356–62.
- Mendler MH, Turlin B, Moirand R, Jouanolle AM, Sapey T, Guyader D, et al. Insulin resistance-associated hepatic iron overload. Gastroenterology 1999;117(5):1155–63.
- Dongiovanni P, Fracanzani AL, Fargion S, Valenti L. Iron in fatty liver and in the metabolic syndrome: a promising therapeutic target. J Hepatol 2011;55(4):920–32.
- George DK, Goldwurm S, MacDonald GA, Cowley LL, Walker NI, Ward PJ, et al. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998;114(2):311–31.
- Sumida Y, Yoshikawa T, Okanoue T. Role of hepatic iron in non-alcoholic steatohepatitis. Hepatol Res 2009;39(3):213–22.
- Fujita N, Miyachi H, Tanaka H, Takeo M, Nakagawa N, Kobayashi Y, et al. Iron overload is associated with hepatic oxidative damage to DNA in nonalcoholic steatohepatitis. Cancer Epidemiol Biomarkers Prev 2009;18(2):424–32.
- Obika M, Noguchi H. Diagnosis and evaluation of nonalcoholic fatty liver disease. Exp Diabetes Res 2012;2012:145754.
- Shyangdan D, Clar C, Ghouri N, Henderson R, Gurung T, Preiss D, et al. Insulin sensitisers in the treatment of non-alcoholic fatty liver disease: a systematic review. Health Technol Assess 2011;15(38):1–110.
- Haukeland JW, Damås JK, Konopski Z, Løberg EM, Haaland T, Goverud I, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol 2006;44(6):1167–74.
- Kadiiska MB, Gladen BC, Baird DD, Germolec D, Graham LB, Parker CE, et al. Biomarkers of oxidative stress study II. Are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radic Biol Med 2005;38(6):698–710.
- Ibrahim MA, Kelleni M, Geddawy A. Nonalcoholic fatty liver disease: current and potential therapies. Life Sci 2013;92(2):114–8.
- Videla LA, Rodrigo R, Araya J, Poniachik J. Oxidative stress and depletion of hepatic long-chain polyunsaturated fatty acids may contribute to nonalcoholic fatty liver disease. Free Radic Biol Med 2004;37(9):1499–507.
- Angulo P. NAFLD, obesity, and bariatric surgery. Gastroenterology 2006;130(6):1848.