Abstract
The prevalence of Alzheimer's disease (AD) is increasing rapidly worldwide due to an ageing population and largely ineffective treatments. In AD cognitive decline is due to progressive neuron loss that begins in the medial temporal lobe and spreads through many brain regions. Despite intense research the pathogenesis of the common sporadic form of AD remains largely unknown. The popular amyloid cascade hypothesis suggests that the accumulation of soluble oligomers of beta amyloid peptides (Aβ) initiates a series of events that cause neuronal loss. Among their putative toxic effects, Aβ oligomers are thought to act as pro-oxidants combining with redox-active metals to produce excessive reactive oxygen and nitrogen species. However, to date the experimental therapies that reduce Aβ load in AD have failed to halt cognitive decline. Another hypothesis proposed by the late Mark Smith and colleagues is that oxidative stress, rather than Aβ, precipitates the pathogenesis of AD. That is, Aβ and microtubule-associated protein tau are upregulated to address the redox imbalance in the AD brain. As the disease progresses, excess Aβ and tau oligomerise to further accelerate the disease process. Here, we discuss redox balance in the human brain and how this balance is affected by ageing. We then discuss where oxidative stress is most likely to act in the disease process and the potential for intervention to reduce its effects.
Introduction
Alzheimer's disease (AD) is the most common form of dementia, accounting for 60% of all cases. The prevalence of AD is approximately 1% of the total population in western countries, while the age-prevalence rises rapidly from 5% at 65 years to greater than 25% in individuals over 85 years of age. The worldwide incidence of AD is increasing dramatically with the 36 million current sufferers worldwide predicted to reach 115 million by 2050.Citation1
The AD brain is characterized by the deposition of insoluble protein as extracellular plaques and intracellular neurofibrillary tangles (NFTs). The former are largely composed of a group of peptides called beta amyloid (Aβ), while the latter are composed of fibrillar and hyperphosphorylated microtubule-associated protein tau (tau). While Aβ and tau deposits are hallmarks of AD and are used for the pathological diagnosis of the disease, dementia results from the regional loss of neurons.
According to the amyloid cascade hypothesis (ACH), oxidative injury is precipitated by the build-up of Aβ oligomers and progresses to tau-mediated neurodegeneration.Citation2 It has been proposed that the recent failure of Aβ-modifying treatments in clinical trials reflects that their implementation at the clinical phase of AD is too late to modify disease progression.Citation3 Opponents of the ACH suggest that a factor X precipitates sporadic forms of AD. In particular, the late Mark Smith and his colleagues suggest that Aβ may act initially as an anti-oxidant that is upregulated in response to oxidative stress (OS).Citation4 Here we review the literature linking OS and AD and define its likely role in this disease.
Clinical presentation
The clinical diagnosis of AD is based on symptoms and made without the aid of sensitive and specific biomarkers. A diagnosis of ‘probable’ AD is made when there is a worsening memory with deficits in two or more other cognitive domains, such as executive function, attention, language, and visuospatial skillsCitation5 in the absence of other conditions likely to cause the dementia.
The diagnostic criteria have recently been revised and now incorporate, at least in the research setting, biomarkers of the underlying pathophysiological process.Citation6 Clinical biomarkers have been divided into those representing ‘early’ Aβ deposition such as can be demonstrated with the Aβ-binding ligand, Citation11C-labelled Pittsburgh Compound-B, positivity on positive emission tomography while ‘late’ events include elevated cerebrospinal fluid (CSF) tau protein and regional brain atrophy.
The preclinical period
The average disease duration of AD is 10 years but the prodrome may be up to 20 years.Citation7 The latter can be divided into an early ‘Preclinical AD’Citation8 and mild cognitive impairment (MCI).Citation9 MCI is distinguished from AD in that the individual does not suffer any significant interference in their ability to function at work or in daily activities.Citation9 The term ‘preclinical AD’ optimistically infers that ‘early’ biomarkers faithfully reflect the underlying pathophysiological processes.Citation8 Indeed, studies suggest that the majority of individuals with MCI have typical AD pathology, but to a lesser degree than those who have dementia.Citation10
The pathogenesis of AD
Aβ accumulation is regarded as the initiating event in AD, but it is the spread of NFT pathology from the medial temporal lobe to the association and finally primary corticesCitation11 that correlates better with the probability and severity of dementia. In comparison, the regional distribution of Aβ plaques varies markedly between patients.Citation11 Similarly, reactive glia, that associate with plaques and NFTs show better correlations with the extent of NFTs than Aβ burden.Citation12
The modern research landscape of AD was established when the Aβ peptide(s) was purified from amyloid-containing AD brain tissue.Citation13 The parent amyloid precursor protein (APP) was subsequently isolated and sequenced from a human brain cDNA library and comparative studies suggested it was a neuronal surface receptor.Citation14 The observation that individuals with trisomy 21 (Down syndrome) develop AD pathology around 40–50 years of ageCitation15 suggested that copy number mutations in APP may account for familial forms of AD. Initially APP missense mutations were found in AD families followed by mutations in two other genes that encode presenilin 1 (PS1) and 2 (PS2) but APP multiplications were also eventually found in 2006.Citation16
APP is degraded via amyloidogenic or non-amyloidogenic pathways. The majority of non-amyloidogenic processing occurs at the plasma membraneCitation17 while the majority of amyloidogenic APP metabolism occurs within the endosomal–lysosomal system following internalization, a process closely related to synaptic activity.Citation18 In the amyloidogenic pathway β- and γ-secretases combine to produce Aβ, a collection of peptides up to 42 amino acids in length.Citation19 The γ-secretase is a multi-unit enzyme that includes either PS1 or PS2.
Although representing fewer than 5% of total AD cases, the discovery that monogenic forms of AD combined with typical AD pathology in Down syndrome led Hardy and Higgins to suggest the ACH for the pathogenesis of sporadic AD.Citation20 The ACH suggests that fibrillar Aβ in plaques disrupts neuronal calcium homeostasis, precipitating abnormal tau hyperphosphorylation, and NFT formation (). The hypothesis was modifiedCitation2 when soluble oligomeric forms of Aβ, that are also referred to as amyloid-beta-derived diffusible ligands (ADDLs), became the more likely pathogenic species.Citation21 The modified hypothesis, which also recognizes the involvement of oxidative injury in the cascade, has stood the test of time although recent failures of Aβ-limiting therapeutics in clinical trials have led some to question an early pivotal role for Aβ in AD.
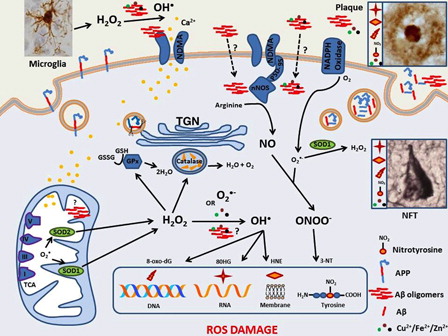
Figure 1. Oxidative stress and Alzheimer's disease: amyloid cascade hypothesis. Accumulation of extracellular soluble beta amyloid (Aβ) oligomers leads to ionic dysregulation, oxidative stress, neurofibrillary tangle formation and finally the death of neurons. Aβ is largely produced in the trans golgi network (TGN) of neurons following internalisation of the amyloid precursor protein (APP) via the endosomal-lysosomal system. Aβ monomers are then transported out of the neuron via the secretory pathway where they may combine with metal ions, such as Cu2+, that precipitate their self-aggregation to oligomers and finally plaques. It is Aβ oligomers that are postulated to disrupt NMDA receptor activity causing an excessive Ca2+ ion influx into adjacent neurons. High cytoplasmic Ca2+ levels disrupt mitochondria leading to decreased efficiency of the electron transport chain and increased superoxide (O2−•) production; O2−• is also generated by the membrane bound NADPH oxidases. O2−• is converted to H2O2 by superoxide dismutases (SOD1 and 2). Reactive microglia are another source of H2O2 and this two-electron oxidant readily diffuses and can be converted to the potent hydroxyl radical (HO•). H2O2 is problematic for neurons if enzymes such as catalase in peroxisomes or cytoplasmic glutathione peroxidase do not adequately degrade it. O2−• can also combine with nitric oxide (NO) to produce peroxynitrite (ONOO−). These reactive oxygen and nitrogen species damage DNA, lipid and proteins resulting in products such as 8-dihydro-2′-guanosine (8OHG), 4-hydroxynonenal (HNE) and 3-nitrotyrosine (3-NT) respectively. All three oxidation products are found in both the extracellular plaques and intraneuronal neurofibrillary tangles that characterize the AD brain. Aβ oligomers may also act as pro-oxidants themselves, upregulate the neuronal form of nitric oxide synthase, or bind and reduce redox active metals allowing them to catalyse the conversion H2O2 to HO• via the Fenton reaction. Extracellular Aβ oligomers are generally thought to precipitate the AD cascade but there is increasing evidence that intraneuronal Aβ accumulation is also an early event in AD and it may accumulate in mitochondria causing direct damage and/or catalyse cytoplasmic redox reactions.
Redox balance and the brain
The brain requires 20% of total blood flow and 25% of the body's glucose utilization. In neurons, energy in the form of adenosine triphosphate (ATP) is produced aerobically through the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OxPhos) in mitochondria. OxPhos is not totally efficient and up to 2% of electrons are incompletely reduced to yield O2−•, instead of H2O ().
Low-molecular weight anti-oxidants such as glutathione, and metal-containing enzymes such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX), act to control the redox environment in the brain.Citation22 The first defence against ROS is SOD converting O2−• to H2O2. Isoforms of SOD include the mitochondrial manganese-SOD (SOD2) and the cytoplasmic copper/zinc SOD (SOD1). CAT and GPX then convert the weak two-electron oxidant H2O2 to oxygen and water. GPX in particular deals with H2O2 and organic peroxidases by catalyzing the conversion of glutathione (GSH) to its oxidized form, glutathione disulphide (GSSG).
Nitric oxide (NO) is a signalling and neuroprotective molecule in the brain but it can combine with O2−• to yield peroxynitrite, a reactive nitrogen species (RNS). Excessive NO competes with the anti-oxidant enzymes for O2−• to promote RNS generation.Citation23
The brain also has relatively high concentrations of redox-active metal ions such as iron (Fe) and copper (Cu) that not only promote ADDL formation but also facilitate the conversion of H2O2 to other potent ROS.Citation24 Therefore, despite being well served by overlapping anti-oxidant defence mechanisms, the redox balance of the brain is permanently challenged, a scenario that worsens with age.
The ageing brain
Ageing is the major risk factor for AD but there is also considerable overlap in the pathology and postulated mechanisms for cognitively normal ageing and AD. The pathognomonic entities of AD, namely NFTs and plaques, are both common in the brains of neurologically normal individuals.Citation25 In fact, tau pathology confined to the locus coeruleus and entorhinal cortex is observed in the vast majority of aged brains.Citation26,Citation27
Importantly, OxPhos not only becomes progressively less efficient with ageingCitation28 but there is a concurrent decrease in anti-oxidant capacity.Citation29 This results in accumulation of ROS that inhibit cellular pathways through damage to lipids, proteins, and DNA.Citation30 Interestingly, treatment of a senescence-accelerated mouse model with the putative anti-oxidant curcumin restored ATP production and mitochondrial membrane potential in brain cells ex vivoCitation31. The close link between mitochondrial dysfunction, ageing and AD hints at a possible role for anti-oxidants in modifying AD progression.
Markers of oxidative stress in AD
The determination of OS is achieved by either measuring reactive oxygen and nitrogen species (RONS) directly or by the secondary damage caused to various macromolecules ().
Proteins can also be targeted directly by RONS or non-specifically by damage to individual amino acids. The TCA cycle enzyme, aconitase, is highly sensitive to inactivation by O2−• because of its labile iron atom and its proximity to the OxPhos. Therefore, a decrease in brain aconitase activity is a sensitive indicator of excess mitochondrial O2−• production. Specifically, O2−• inactivates aconitase by displacing Fe2+from the Fe-S cluster, which is then available to react with H2O2 to generate secondary ROS (e.g. HO• and ROO•). Tyrosine is nitrated by peroxynitrite to 3-nitrotyrosine, not only disrupting function but also acting as a marker for excess peroxynitrite.Citation32 Redox sensitive amino acid residues can also be oxidized by ROS to yield carbonyl derivatives and disulphide bonds at cysteine residues that result in structural and functional damage to proteins.Citation33 Lastly, advanced glycation end products resulting from condensation of reducing sugars with proteins via the Maillard reaction are also often oxidized, further compromising the protein's functional activity.Citation34
Oxidative damage to DNA and RNA produces modified bases such as 8-oxo-2′-deoxyguanosine (8-oxo-dG) and 8-dihydro-2′-guanosine (8OHG), respectively, and both have been reported in cerebral homogenates derived from AD brains.Citation35 Structural alterations in DNA can have functional consequences such as base pair mismatching and changes in DNA repair mechanisms.Citation36 RNA oxidation leads to decreased protein synthesis when damaged transcripts are degraded by RNA surveillance systems or rejected by translation machinery.Citation37
The peroxidation of membranous polyunsaturated fatty acids by ROS yield products such as iso- and neuro-prostanes, acrolein, and the reactive aldehydes, malondialdehyde and 4-hydroxy-2-nonenal (HNE), and these are all found in the AD brain.Citation36 Lipid oxidation products can act directly in the cell or, in the case of HNE, covalently attach to proteins to inhibit their functions. HNE-conjugated proteins, α-enolase and voltage-dependent anion channel 1, are not only seen in AD brain tissue but also Huntington's disease and amyotrophic lateral sclerosis.Citation38 Similarly, increased acrolein and HNE levels have been described in post-mortem brain tissues of patients with MCICitation39 while CSF isoprostanes have also been described in offspring of female AD patients.Citation40 The latter study argues for perturbations in redox balance occurring in individuals with a potential genetic susceptibility to AD.
The mechanism of oxidative stress in AD
In 1994 the Butterfield group demonstrated in vitro that Aβ was a pro-oxidantCitation41 and that oxidation of the sulphur atom of methionine residue 35 in Aβ1–42 or shorter fragments was critical to its pro-oxidant and neurotoxic properties.Citation42 It was suggested that Cu2+converts Aβ to a sulphoperoxy radical.Citation43 As a variation on this theme, Greenough et al.Citation24 consider that it is the binding of the redox-active metals Cu and Fe to Aβ that facilitates their reduction and the consequent pro-oxidant activity of these Aβ-metal ion complexes ().
Soluble Aβ also has been shown to stimulate neuronal nitric oxide synthetase (nNOS) to produce NO, which can then combine with O2−• to produce peroxynitrite.Citation44 However, nNOS is also activated by NMDA receptor-mediated calcium influxCitation45 so NO-mediated oxidative damage is not necessarily due to the direct effects of Aβ.
In addition, Aβ is also thought to damage mitochondria, either indirectly via excessive intraneuronal Ca2+ levels or directly by acting as a mitochondrial toxin.Citation42 A proteomic study utilizing the triple transgenic (APP/PS1/tau) mouse showed deficits in OxPhos with an Aβ-dependent inhibition of the electron transport chain complex IV but a tau-dependent inhibition of complex I.Citation46Aβ is known to accumulate intraneuronallyCitation47 and it may be taken up by mitochondria, although it seems more likely that putative interactions involve binding to mitochondrial surface proteins such as amyloid-binding alcohol dehydrogenase.Citation48
Metals and cerebral oxidative stress
Several transition metals play key roles in brain metabolism, including Fe, Cu, Zinc (Zn), and manganese, but in excess they may be involved in chemical reactions that form ROS including H2O2, O2−• and HO•. Studies of post-mortem brain tissue showed that CuCitation49 and FeCitation50 were both elevated in the AD brain. However, more recently this metal hypothesis of AD was questioned when a meta-analysis suggested that there were no global increases in these metals; Zn and Fe concentrations remained unchanged while Cu was actually depleted in the AD brain.Citation51 Proponents of the metal hypothesis of AD countered by suggesting that there were focal increases in metal content around plaques and NFTs.Citation24
It is thought that haemoxygenase (HO) provides an important source of redox-active Fe2+ that must be sequestered in a ‘redox-inactive’ form.Citation52 As part of normal homeostasis, constitutively expressed HO-2 regulates pro-oxidant haem by degrading it into free Fe, carbon monoxide and biliverdin/bilirubin. However, when confronted with increased levels of haem, inducible HO-1 is produced and this often occurs in parallel with increases in the intracellular iron-binding protein ferritin, which binds the released Fe.Citation53 Potentially, free haem can cause oxidation of other proteins and lipids.Citation54 Similarly, unbound Fe2+ can react with H2O2 to yield highly reactive OH• (Fenton's reaction) or interact with O2−• and H2O2 to form OH• via the Haber–Weiss reaction (). Interestingly, HO-1 localizes with NFTs in the AD brain.Citation55 Accumulating Fe2+ and Cu2+ ions can bind to histidyl and sulphydryl residues, respectively, within Aβ.Citation24 These Aβ-metal complexes are also able to generate ROS via Haber–Weiss and Fenton reactions.
One limitation in AD research is the lack of understanding of the physiological role of APP. We do know that levels of Zn, Cu, and Fe can all affect APP metabolism.Citation56 The APP gene contains a Fe-responsive element in its 5′ untranslated regionCitation57 and Fe regulatory proteins are able to control its translation. APP also contains a Cu-binding site and can reciprocally modulate the levels of Cu2+in the brain.Citation58 Both APP and Aβ may be involved in maintaining metal homeostasis in the brain, particularly in the face of increased OS.Citation59
Anti-oxidant enzymes
The data from post-mortem AD brains are inconsistent in terms of the levels of anti-oxidant enzymes including SOD1 or total SOD activity, with increases,Citation60 decreases,Citation61 and no differencesCitation62 found compared to controls. GPX levels have been reported as decreased or showing no differenceCitation63 with only one report of increased GPX in the AD brain.Citation64 CAT levels have also been reported as either decreasedCitation65 or not different.Citation66
Karelson et al. showed that SOD activity was highly correlated with plaques but not NFTsCitation60 but Maeda et al.Citation67 suggested that this was mainly SOD2 rather than SOD1. Like APP, SOD1 is located on chromosome 21 and SOD1 is overexpressed in Down syndrome brains.Citation68 It seems paradoxical that the anti-oxidant enzyme SOD could be linked with neurodegeneration but the generation of H2O2 is potentially toxic for the cell if enzymes such as CAT or GPX are compromised. Consistent with this scenario, Down syndrome neurons can be rescued by CAT expressionCitation69 while neurotoxic effects of Aβ on hippocampal neurons can be ameliorated by the combination of CAT and SOD expression.Citation70 Similarly, it was recently shown that a mitochondrial-targeted CAT/APP double transgenic mouse displayed reduced APP processing and Aβ load with enhanced Aβ-degrading enzyme activity compared to the APP single transgenic.Citation71
An alternative hypothesis
Results from the CAT/APP mouse model suggest that ROS, and particularly H2O2, can induce the upregulation of Aβ rather than the other way around. This view was held by the late Mark Smith and has been summarized in a recent review article.Citation4 Briefly, Cuajungco et al.Citation72 consider that neurons respond to OS by increasing Aβ production, with Aβ acting as a SOD mimic. Furthermore, Aβ, by binding metal ions, may prevent them from participating in redox cycling reactions.Citation73
The group also maintain that tau is upregulated in response to OS, based firstly on neurons with NFTs in AD brains having less 8-OHG and 3-NTCitation74 with similar findings in Down syndromeCitation75 and monogenic AD patients.Citation76 Second, the phospho-protein tau and other structural proteins act as physiological ‘sponges’ for oxidation products such as toxic aldehydes.Citation77 Smith et al.Citation78 suggest that plaques and NFTs in elderly controls are likely to reflect increased levels of OS associated with the aged brain and similarly question whether the same metal-laden entities are the source of OS in AD. In contrast, they favour the idea that intraneuronal Aβ is more important and accumulates as a cellular response to OS. Dysfunctional mitochondria are the likely source of ROS because HO•, which diffuses over nanometre distances only, must be generated in the cytosol in intimate proximity to RNA to yield 8OHG ().Citation75 In addition, 8OHG levels correlate with the onset of cognitive impairment in the prodromal stage of AD and are greater than that seen in normal ageing.Citation79 As excessive Aβ and tau are neurotoxic, their initial upregulation is likely to provide only transient relief. Such a scenario would still be consistent with the genetic evidence in AD, considering that most monogenic forms of AD manifest by middle age.Citation80
Testing the oxidative stress hypothesis
Presently the only therapeutics approved for use in AD are the acetylcholinesterase inhibitors and memantine, a NMDA-receptor antagonist. While offering transient improvements in cognition these therapies do not slow disease progression and disease-modifying treatments are sorely needed. Direct evidence for the OS/free radical hypothesis of AD would include a demonstrable ability of anti-oxidants to modify disease course. In this respect the overall results have been disappointing.Citation81 Proponents suggest that the variety of trial designs and anti-oxidants used make meta-analyses difficult and that it is still too early to discount a role for anti-oxidants in AD treatment. For example, a trial of vitamin C in an APP double mutant mouse yielded a subtle decrease in Aβ soluble oligomers and protein carbonylation, increased glutathione levels and improved the behavioural phenotype with no amyloid deposition or reactive gliosis.Citation82
A recent trial looked at the anti-oxidant combination of vitamins E, C and α-lipoic acid or coenzyme Q in AD patients.Citation83 This trial was novel in that it assayed biomarkers of OS in the CSF. Although there was no effect of treatment on CSF biomarkers related to amyloid or tau pathology, F2-isoprostane levels decreased in subjects receiving vitamins E, C, and α-lipoic acid, although not coenzyme Q. Unfortunately, F2-isoprostane concentration also correlated with decreasing cognitive performance on a Mini-Mental State Examination (MMSE). Moreover, a Cochrane Database Systematic Review for vitamin ECitation84 found no convincing evidence of benefit in treating AD or MCI. Possibly, vitamin E may only inhibit lipid peroxidation with no effects on the oxidative damage to proteins and nucleic acids.Citation34 A review of vitamin C supplementation in AD suggests that avoiding vitamin C deficiency via a balanced diet is more important than supplementation in AD prevention.Citation85
Given the lack of clinical success of anti-oxidant supplements, research has refocussed on limiting the production of ROS such as modulating NAPDH oxidase activity.Citation86 Recent studies have also suggested that selenium, the metal co-factor for GPX, might be deficient in AD.Citation87 One report in an AD mouse model suggested that selenium supplementation can attenuate tau pathology, but it appeared to stabilize a complex formed between tau and its major protein phosphatase, PP2A,Citation88 rather than augmenting GPX activity.
One area where some clinical success has been reported is the trials with metal chelators. As noted Cu, Fe, and Zn can bind to Aβ and precipitate aggregation while Cu or Fe can form complexes with Aβ that enhance H2O2 production. The first metal chelator trialled, clioquinol, had initial encouraging results in phase II studies;Citation89 however, no improvement in cognitive performance could be established and the phase II trial was abandoned.Citation90 A clioquinol-like drug, PBT2 was subsequently developed and it performed well in a phase IIa trial with improvements in cognition on some neuropsychological tests.Citation91 However, a Cochrane Review in 2012 noted that no improvements in cognition were seen with either the MMSE or ADAS-Cog scale.Citation92 The current PBT2 phase II trial is due for completion in December 2013.
Conclusion
The working model for the pathogenesis of sporadic AD is the ACH, which states that the accumulation of soluble Aβ oligomers has various deleterious effects on neurons, including oxidative injury, that eventually result in tau-mediated death. An alternative (free radical) hypothesis suggests that OS precedes Aβ oligomerization and that Aβ and tau are upregulated because they can act as anti-oxidants. The latter hypothesis is certainly not as popular but is supported by reasonable evidence. However, it is also possible over the long preclinical and clinical phases of AD that both hypotheses could be operative with Aβ and OS combining to cause cellular damage or that Aβ and OS alternate in their pathogenic roles as the brain attempts to compensate for the disease process.
Understanding the different stages of AD is relevant to treatment regimens, likely preclinical markers and outcome. At present diagnoses are made on clinical signs that result from neuronal loss. The mechanisms responsible for cell loss at the clinical stage of AD might be quite different to those operating in early degenerating neurons. The continuing failure of the very promising Aβ-modifying therapies may be related to these therapies being administered too late, and/or other factors being the precipitating driver of AD pathogenesis. If that driver is OS, then there is probably less chance that anti-oxidants will be therapeutically beneficial if only used in clinical cases as the available data suggest.
In reality the predictable spread of AD pathology across the brain means that neurons of different regions will be at different stages of degeneration. Therefore, therapy is likely to require a suite of therapeutics that target ‘preclinical’ and ‘clinical’ pathogenic mechanisms. One caveat here is that a protective host response in one region could have reached deleterious proportions elsewhere. It is also possible that we are yet to sufficiently understand the physiological roles of APP, Aβ, and ROS to predict the chain of events in AD. Clinical trials based on our most likely therapeutic targets are an essential avenue to prevent an AD epidemic, but similar enthusiasm is required to tease apart the pathogenesis of this most complex of diseases.
Acknowledgements
A Judith Jane Mason and Harold Stannett Williams Memorial Foundation Medical and Scientific Research grant supported this work. The authors thank Mr Stephen Kum Jew and Mr Claude Dennis for their assistance with immunohistochemistry and Professor Jillian Kril for her useful comments and suggestions.
Related Research Data
References
- Alzheimer's Disease International. World Alzheimer Report 2010. London; 2009.
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297(5580):353–6.
- Golde TE, Schneider LS, Koo EH. Anti-abeta therapeutics in Alzheimer's disease: the need for a paradigm shift. Neuron 2011;69(2):203–13.
- Swerdlow RH. Alzheimer's disease pathologic cascades: who comes first, what drives what. Neurotox Res 2012;22(3):182–94.
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34(7):939–44.
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR , Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7(3):263–9.
- Peskind ER, Li G, Shofer J, Quinn JF, Kaye JA, Clark CM, et al. Age and apolipoprotein E*4 allele effects on cerebrospinal fluid beta-amyloid 42 in adults with normal cognition. Arch Neurol 2006;63(7):936–9.
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7(3):280–92.
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7(3):270–9.
- Haroutunian V, Hoffman LB, Beeri MS. Is there a neuropathology difference between mild cognitive impairment and dementia? Dialogues Clin Neurosc 2009;11(2):171–9.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82(4):239–59.
- Serrano-Pozo A, Mielke ML, Gomez-Isla T, Betensky RA, Growdon JH, Frosch MP, et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol 2011;179(3):1373–84.
- Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 1984;122(3):1131–5.
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987;325(6106):733–6.
- Seidl R, Cairns N, Lubec G. The brain in Down syndrome. J Neural Transm Suppl 2001;61:247–61.
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 2006;38(1):24–6.
- Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harbor Perspect Med 2012;2(5):a006270.
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, et al. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci 2011;14(6):750–6.
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci 2007;8(7):499–509.
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992;256(5054):184–5.
- Shankar GM, Walsh DM. Alzheimer's disease: synaptic dysfunction and Abeta. Mol Neurodegener 2009;4:48.
- von Bernhardi R, Eugenin J. Alzheimer's disease: redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid Redox Signal 2012;16(9):974–1031.
- Bishop A, Anderson JE. NO signaling in the CNS: from the physiological to the pathological. Toxicology 2005;208(2):193–205.
- Greenough MA, Camakaris J, Bush AI. Metal dyshomeostasis and oxidative stress in Alzheimer's disease. Neurochem Int 2013;62(5):540–55.
- Davis DG, Schmitt FA, Wekstein DR, Markesbery WR. Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol 1999;58(4):376–88.
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997;18(4):351–7.
- Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol 2011;121(2):171–81.
- Marcinek DJ, Schenkman KA, Ciesielski WA, Lee D, Conley KE. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J Physiol 2005;569(Pt 2):467–73.
- Jones DP. Extracellular redox state: refining the definition of oxidative stress in aging. Rejuvenation Research 2006;9(2):169–81.
- Kowald A. The mitochondrial theory of aging. Biol Signals Recept 2001;10(3–4):162–75.
- Eckert GP, Schiborr C, Hagl S, Abdel-Kader R, Muller WE, Rimbach G, et al. Curcumin prevents mitochondrial dysfunction in the brain of the senescence-accelerated mouse-prone 8. Neurochem Int 2013;62(5):595–602.
- Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Evidence of neuronal oxidative damage in Alzheimer's disease. Am J Pathol 1996;149(1):21–8.
- Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003;25(3–4):207–18.
- Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med 1997;23(1):134–47.
- Nunomura A, Honda K, Takeda A, Hirai K, Zhu X, Smith MA, et al. Oxidative damage to RNA in neurodegenerative diseases. J Biomed Biotechnol 2006;2006(3):82323.
- Sultana R, Butterfield DA. Role of oxidative stress in the progression of Alzheimer's disease. J Alzheimers Dis 2010;19(1):341–53.
- Li Z, Wu J, Deleo CJ. RNA damage and surveillance under oxidative stress. IUBMB Life 2006;58(10):581–8.
- Reed TT. Lipid peroxidation and neurodegenerative disease. Free Radic Biol Med 2011;51(7):1302–19.
- Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease. Neurobiol Aging 2006;27(8):1094–9.
- Mosconi L, Glodzik L, Mistur R, McHugh P, Rich KE, Javier E, et al. Oxidative stress and amyloid-beta pathology in normal individuals with a maternal history of Alzheimer's. Biol Psychiatry 2010;68(10):913–21.
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, et al. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA 1994;91(8):3270–4.
- Butterfield DA. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res 2002;36(12):1307–13.
- Butterfield DA, Bush AI. Alzheimer's amyloid beta-peptide (1–42): involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiol Aging 2004;25(5):563–8.
- Hu J, Akama KT, Krafft GA, Chromy BA, Van Eldik LJ. Amyloid-beta peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res 1998;785(2):195–206.
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Ann Rev Physiol 1995;57:683–706.
- Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc Natl Acad Sci USA 2009;106(47):20057–62.
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, et al. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 2002;161(5):1869–79.
- Borger E, Aitken L, Muirhead KE, Allen ZE, Ainge JA, Conway SJ, et al. Mitochondrial beta-amyloid in Alzheimer's disease. Biochem Soc Trans 2011;39(4):868–73.
- Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, et al. Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem 1999;274(52):37111–6.
- Rottkamp CA, Raina AK, Zhu X, Gaier E, Bush AI, Atwood CS, et al. Redox-active iron mediates amyloid-beta toxicity. Free Radic Biol Med 2001;30(4):447–50.
- Schrag M, Mueller C, Oyoyo U, Smith MA, Kirsch WM. Iron, zinc and copper in the Alzheimer's disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog Neurobiol 2011;94(3):296–306.
- Moreira PI, Sayre LM, Zhu X, Nunomura A, Smith MA, Perry G. Detection and localization of markers of oxidative stress by in situ methods: application in the study of Alzheimer disease. Methods Mol Biol 2010;610:419–34.
- Vile GF, Tyrrell RM. Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. J Biol Chem 1993;268(20):14678–81.
- Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicol Lett 2005;157(3):175–88.
- Smith MA, Kutty RK, Richey PL, Yan SD, Stern D, Chader GJ, et al. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer's disease. Am J Pathol 1994;145(1):42–7.
- Ayton S, Lei P, Bush AI. Metallostasis in Alzheimer's disease. Free Radic Biol Med. 2012 Nov 9. pii: S0891-5849(12)01800-X. doi: 10.1016/j.freeradbiomed.2012.10.558. [Epub ahead of print].
- Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer's amyloid precursor protein transcript. J Biol Chem 2002;277(47):45518–28.
- Acevedo KM, Hung YH, Dalziel AH, Li QX, Laughton K, Wikhe K, et al. Copper promotes the trafficking of the amyloid precursor protein. J Biol Chem 2011;286(10):8252–62.
- Avramovich-Tirosh Y, Amit T, Bar-Am O, Weinreb O, Youdim MB. Physiological and pathological aspects of Abeta in iron homeostasis via 5′UTR in the APP mRNA and the therapeutic use of iron-chelators. BMC Neurosci 2008;9 (Suppl 2):S2.
- Karelson E, Bogdanovic N, Garlind A, Winblad B, Zilmer K, Kullisaar T, et al. The cerebrocortical areas in normal brain aging and in Alzheimer's disease: noticeable differences in the lipid peroxidation level and in antioxidant defense. Neurochem Res 2001;26(4):353–61.
- Ramassamy C, Averill D, Beffert U, Bastianetto S, Theroux L, Lussier-Cacan S, et al. Oxidative damage and protection by antioxidants in the frontal cortex of Alzheimer's disease is related to the apolipoprotein E genotype. Free Radic Biol Med 1999;27(5–6):544–53.
- Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol 2010;69(2):155–67.
- Omar RA, Chyan YJ, Andorn AC, Poeggeler B, Robakis NK, Pappolla MA. Increased expression but reduced activity of antioxidant enzymes in Alzheimer's disease. J Alzheimers Dis 1999;1(3):139–45.
- Schuessel K, Leutner S, Cairns NJ, Muller WE, Eckert A. Impact of gender on upregulation of antioxidant defence mechanisms in Alzheimer's disease brain. J Neural Transm 2004;111(9):1167–82.
- Casado A, Encarnacion Lopez-Fernandez M, Concepcion Casado M, de La Torre R. Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem Res 2008;33(3):450–8.
- Chen L, Richardson JS, Caldwell JE, Ang LC. Regional brain activity of free radical defense enzymes in autopsy samples from patients with Alzheimer's disease and from nondemented controls. Int J Neurosci 1994;75(1–2):83–90.
- Maeda M, Takagi H, Hattori H, Matsuzaki T. Localization of manganese superoxide dismutase in the cerebral cortex and hippocampus of Alzheimer-type senile dementia. Osaka City Med J 1997;43(1):1–5.
- Gulesserian T, Seidl R, Hardmeier R, Cairns N, Lubec G. Superoxide dismutase SOD1, encoded on chromosome 21, but not SOD2 is overexpressed in brains of patients with Down syndrome. J Investig Med 2001;49(1):41–6.
- Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro. Nature 1995;378(6559):776–9.
- Aksenov MY, Aksenova MV, Markesbery WR, Butterfield DA. Amyloid beta-peptide (1–40)-mediated oxidative stress in cultured hippocampal neurons. Protein carbonyl formation, CK BB expression, and the level of Cu, Zn, and Mn SOD mRNA. J Mol Neurosci 1998;10(3):181–92.
- Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, Reddy AP, et al. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer's disease: implications for neuroprotection and lifespan extension. Hum Mol Genet 2012;21(13):2973–90.
- Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, et al. Evidence that the beta-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem 2000;275(26):19439–42.
- Atwood CS, Obrenovich ME, Liu T, Chan H, Perry G, Smith MA, et al. Amyloid-beta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Brain Res Brain Res Rev 2003;43(1):1–16.
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 2001;60(8):759–67.
- Nunomura A, Perry G, Hirai K, Aliev G, Takeda A, Chiba S, et al. Neuronal RNA oxidation in Alzheimer's disease and Down's syndrome. Ann N Y Acad Sci 1999;893:362–4.
- Nunomura A, Chiba S, Lippa CF, Cras P, Kalaria RN, Takeda A, et al. Neuronal RNA oxidation is a prominent feature of familial Alzheimer's disease. Neurobiol Dis 2004;17(1):108–13.
- Wataya T, Nunomura A, Smith MA, Siedlak SL, Harris PL, Shimohama S, et al. High molecular weight neurofilament proteins are physiological substrates of adduction by the lipid peroxidation product hydroxynonenal. J Biol Chem 2002;277(7):4644–8.
- Nunomura A, Tamaoki T, Tanaka K, Motohashi N, Nakamura M, Hayashi T, et al. Intraneuronal amyloid beta accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol Dis 2010;37(3):731–7.
- Nunomura A, Tamaoki T, Motohashi N, Nakamura M, McKeel DW, Tabaton M, et al. The earliest stage of cognitive impairment in transition from normal aging to Alzheimer disease is marked by prominent RNA oxidation in vulnerable neurons. J Neuropathol Exp Neurol 2012;71(3):233–41.
- Joshi A, Ringman JM, Lee AS, Juarez KO, Mendez MF. Comparison of clinical characteristics between familial and non-familial early onset Alzheimer's disease. J Neurol 2012;259(10):2182–8.
- Teixeira J, Silva T, Andrade PB, Borges F. Alzheimer Disease and Antioxidant Therapy: how long how far? Curr Med Chem 2013 Jan 28 [Epub ahead of print].
- Murakami K, Murata N, Noda Y, Tahara S, Kaneko T, Kinoshita N, et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J Biol Chem 2011;286(52):44557–68.
- Galasko DR, Peskind E, Clark CM, Quinn JF, Ringman JM, Jicha GA, et al. Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol 2012;69(7):836–41.
- Farina N, Isaac MG, Clark AR, Rusted J, Tabet N. Vitamin E for Alzheimer's dementia and mild cognitive impairment. Cochrane Database Syst Rev 2012;11:CD002854.
- Harrison FE. A critical review of vitamin C for the prevention of age-related cognitive decline and Alzheimer's disease. J Alzheimers Dis 2012;29(4):711–26.
- Gao HM, Zhou H, Hong JS. NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol Sci 2012;33(6):295–303.
- Loef M, Schrauzer GN, Walach H. Selenium and Alzheimer's disease: a systematic review. J Alzheimers Dis 2011;26(1):81–104.
- van Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Gotz J, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer's disease models. Proc Natl Acad Sci USA 2010;107(31):13888–93.
- Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol 2003;60(12):1685–91.
- Sampson E, Jenagaratnam L, McShane R. Metal protein attenuating compounds for the treatment of Alzheimer's disease. Cochrane Database Syst Rev. 2008 Jan 23;(1):CD005380. doi: 10.1002/14651858.CD005380.pub3.
- Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, et al. PBT2 rapidly improves cognition in Alzheimer's Disease: additional phase II analyses. J Alzheimers Dis 2010;20(2):509–16.
- Sampson EL, Jenagaratnam L, McShane R. Metal protein attenuating compounds for the treatment of Alzheimer's dementia. Cochrane Database Syst Rev 2012;5:CD005380.