Abstract
Oxidative stress plays a role in a variety of diseases but it is even more pertinent in chronic obstructive pulmonary disease (COPD) given the increased oxidant burden in smokers. The increased oxidant burden results from the fact that cigarette smoke contains over 4700 different chemical compounds and more than 1015 oxidants/free radicals per puff. Other factors, such as air pollutants, infections, and occupational dusts that may exacerbate COPD, also have the potential to produce oxidative stress. These oxidants give rise to Reactive Oxygen Species (ROS) that are generated enzymatically by inflammatory and epithelial cells within the lung as part of an inflammatory immune response towards a pathogen or irritant. Thus, while ROS are necessary for host defence against invading pathogens, increased levels of ROS have been implicated in initiating inflammatory responses in the lungs through the activation of transcriptional factors, signal transduction pathways, chromatin remodelling and gene expression of pro-inflammatory mediators. However, the normal lung has developed defences to ROS-mediated damage, which include antioxidant enzymes such as superoxide dismutase, catalase, and glutathione peroxidase. In this review we consider the therapeutic potential of the antioxidant enzyme glutathione peroxidase-1 for the treatment of cigarette smoke-induced lung inflammation and damage.
Introduction
Cigarette smoking is the major cause of chronic obstructive pulmonary disease (COPD) and accounts for more than 95% of cases in industrialized countries, but other environmental pollutants are important causes in developing countries.Citation1 COPD is ‘a disease state characterized by airflow limitation that is not fully reversible. The airflow limitation is usually progressive and associated with an abnormal inflammatory response of lungs to noxious particles and gases’.Citation1 COPD encompasses chronic obstructive bronchiolitis with fibrosis and obstruction of small airways, and emphysema with enlargement of airspaces and destruction of lung parenchyma, loss of lung elasticity, and closure of small airways. Most patients with COPD have all three pathological conditions (chronic obstructive bronchiolitis, emphysema, and mucus plugging), but the relative extent of emphysema and obstructive bronchiolitis within individual patients can vary.
It is well established that macrophages, neutrophils and lymphocytes are all involved in the pathogenesis of COPD.Citation1 However, several studies have highlighted that macrophages play a pivotal role in the pathophysiology of COPD and can account for most of the known features of the disease.Citation2 There is a marked increase in the numbers of macrophages in airways, lung parenchyma, bronchoalveolar lavage fluid (BALF) and sputum in patients with COPD.Citation3–Citation5 There is a correlation between macrophage numbers in the airways and the severity of COPD.Citation6 Macrophages are activated by cigarette smoke and other irritants to release inflammatory mediators including tumour necrosis factor (TNF), monocyte chemotactic peptide (MCP)-1, reactive oxygen species (ROS) and neutrophil chemotactic factors such as leukotriene B4 and interleukin (IL)-8.Citation1 It is now clear from mouse models of lung inflammation that different macrophage subpopulations exist in the inflamed lung and that although the existence of such subpopulations is implicated in COPD, the importance of these subpopulations is unknown.Citation7 Inflammatory macrophages recruited from circulating monocytes are phenotypically different from the resident population of cells and secrete many cytokines important in pro-inflammatory responses.Citation7 Alveolar macrophages also secrete elastolytic enzymes, including matrix metalloprotease (MMP)-2, MMP-9 and MMP-12, and cathepsin K, L and S which together are responsible for destruction of lung parenchyma.Citation1 In patients with emphysema there is an increase in BALF concentrations and macrophage expression of MMP-1 and MMP-9Citation8 and an increase in activity of MMP-9 in the lung parenchyma.Citation9 Alveolar macrophages from normal smokers express more MMP-9 than those from normal subjectsCitation10 and there is an even greater increase in cells from patients with COPD, which have enhanced elastolytic activity.Citation11
Histological and bronchial biopsy studies show that patients with COPD have an increased number of neutrophils.Citation5,Citation6 In addition, BALF and sputum of COPD patients have a marked increase in neutrophils.Citation3,Citation4 Neutrophil numbers in bronchial biopsies and induced sputum are correlated with COPD disease severity and with the rate of decline in lung function.Citation4,Citation6 Neutrophils secrete serine proteases, including neutrophil elastase, cathepsin G, and protease-3, as well as MMP-8 and MMP-9, which contribute to alveolar destruction and produce emphysema in laboratory animals.Citation1 Neutrophils migrate into the respiratory tract under the direction of neutrophil chemotactic factors.Citation1
Oxidative stress in the pathogenesis of COPD
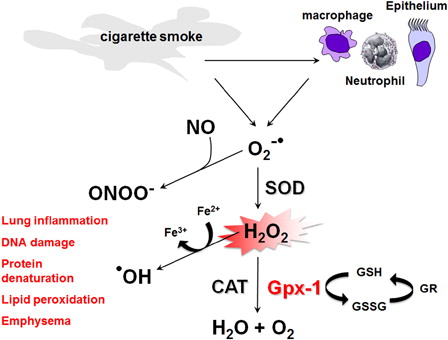
Oxidative stress plays a role in a variety of diseases but it is even more pertinent in COPD given the increased oxidant burden in smokers. The increased oxidant burden results from the fact that cigarette smoke contains over 4700 different chemical compounds and more than 1015 oxidants/free radicals per puff.Citation12–Citation15 Other factors, such as air pollutants, infections, and occupational dusts that may exacerbate COPD, also have the potential to produce oxidative stress. These oxidants give rise to ROS, which are a family of highly reactive molecules that are generated enzymatically by inflammatory and epithelial cells within the lung as part of an inflammatory immune response towards a pathogen or irritant. Activation of macrophages and neutrophils by cigarette smoke generates superoxide radicals (O2•−), which can then either react with nitric oxide (NO) to form reactive peroxynitrite molecules (ONOO−) or alternatively be rapidly converted to damaging hydrogen peroxide (H2O2) under the influence of superoxide dismutase (SOD). This in turn can result in the non-enzymatic production of the more damaging hydroxyl radical (•OH) from H2O2 in the presence of Fe2+ through the Fenton reaction (). This leads to the oxidation of Fe2+ and Fe3+, which in turn can also generate •OH direct from O2•− and regenerate Fe2+ through the Haber-Weiss reaction. Thus, while ROS are necessary for host defence against invading pathogens, increased levels of ROS have been implicated in initiating inflammatory responses in the lungs through the activation of transcriptional factors such as nuclear factor kappaB (NFκB) and activator protein-1 (AP-1), signal transduction pathways, chromatin remodelling, and gene expression of pro-inflammatory mediators.Citation12,Citation16 However, the normal lung has developed defences to ROS-mediated damage, which include the antioxidant enzymes SOD, catalase and glutathione peroxidase (Gpx).
Figure 1. Sources and production of ROS in the lung. Activation of macrophages, neutrophils, and epithelium by cigarette smoke generates superoxide radicals (O2•−) which can then either react with nitric oxide (NO) to form reactive peroxynitrite molecules (ONOO−) or alternatively be rapidly converted to damaging hydrogen peroxide (H2O2) under the influence of SOD. This in turn can result in the non-enzymatic production of damaging hydroxyl radical (•OH) from H2O2 in the presence of Fe2+. H2O2 is subsequently enzymatically reduced by glutathione peroxidases (Gpxs), including Gpx-1, as well as catalase (CAT). Gpx-1 uses GSH as a cofactor to reduce H2O2, resulting in the formation of oxidized glutathione (GSSG), which can then be reduced to GSH by glutathione reductase (GR). The ROS O2•−, ONOO−, H2O2, and •OH can then cause lung inflammation, DNA damage, protein denaturation, lipid peroxidation, and emphysema.
Sources of ROS in the lung
The lungs are exposed to essentially two sources of ROS, environmental and cellular. Environmental-derived ROS consists of both gaseous and particulate air pollution. This ranges from cigarette smoke and oxidant gases, such as ozone, nitrogen dioxide and sulphur dioxide, to airborne particulate matter <10 µM (PM10) from diesel car exhaust fumes that can promote ROS production in situ. The nature of ROS found within cigarette smoke varies from short-lived oxidants, such as O2•− and NO, to long lived organic radicals, such as semiquinones that can undergo redox cycling within the epithelial lining fluid of smokers for some considerable period of time.Citation12,Citation16
Cellular-derived ROS are enzymatically produced by inflammatory and epithelial cells within the lung as part of an inflammatory-immune response towards a pathogen or irritant. Several sources for ROS production exist within a cell and include mitochondrial respiration, NADPH oxidase, and the xanthine/xanthine oxidase system, of which the principal ROS generator is NADPH oxidase. Oxidants present in cigarette smoke can stimulate alveolar macrophages to produce ROS and to release a host of mediators, some of which attract neutrophils and other inflammatory cells into the lungs. Both macrophages and neutrophils, which are known to migrate in increased numbers into the lungs of people with COPD, can generate ROS via the NADPH oxidase system.Citation17 Alveolar macrophages obtained in BAL fluid from the lungs of smokers are more activated to release ROS compared with those obtained from non-smokers.Citation17 One manifestation of this is the release of increased amounts of ROS such as O2•− and H2O2.Citation17 Exposure to cigarette smoke in vitro also has been shown to increase the oxidative metabolism of alveolar macrophages.Citation18 Subpopulations of alveolar macrophages with a higher granular density appear to be more prevalent in the lungs of smokers and are responsible for the increased O2•− production associated with macrophages from smokers.Citation18,Citation19 The generation of ROS in epithelial lining fluid may be further enhanced by the presence of increased amounts of free iron in the pulmonary airspaces in smokers.Citation20,Citation21 This is relevant to COPD since the intracellular iron content of alveolar macrophages is augmented in cigarette smokers and is further increased in those who develop chronic bronchitis, compared with non-smokers.Citation22 In addition, macrophages obtained from smokers release more free iron in vitro than those from non-smokers.Citation23 Not surprisingly, the net effect of all these ROS activities is that smokers and patients with COPD have higher levels of exhaled H2O2 than non-smokers,Citation24 and levels are even higher during exacerbations of COPD.Citation25 This increase in H2O2 is in part derived from increased release of O2•− from alveolar macrophages in smokers.Citation25 H2O2 can induce apoptosis in airway epithelial cellsCitation26 and evidence from studies both in animals and man has shown that apoptosis occurs in smoke-exposed macrophages and airway epithelial cells.Citation27–Citation29
Macrophages also employ other enzymes to produce ROS. This involves the activity of the heme peroxidase, myeloperoxidase, or eosinophil peroxidase, which are also found in neutrophils and eosinophils, respectively. These enzymes catalyse the formation of the potent and very damaging oxidants hypochlorous acid (HOCl) and hypobromous acid (HOBr) from H2O2 in the presence of chloride (Cl−) and bromide (Br−) ions, respectively. Circulating neutrophils from cigarette smokers and patients with exacerbations of COPD release more O2•−.Citation30 In addition, an association between O2•− release by peripheral blood neutrophils and bronchial hyperreactivity in patients with COPD has been shown, suggesting a role for systemic ROS in the pathogenesis of COPD.Citation31
When generated close to cell membranes, ROS oxidize membrane phospholipids (lipid peroxidation), a process that may continue as a chain reaction, generating many lipid hydroperoxide molecules within the cell membrane.Citation32 This can result in the oxidation of proteins, DNA, and lipids that may cause direct tissue injury or induce a variety of cellular responses, through the generation of secondary metabolic reactive species. This can impair membrane function, inactivate membrane-bound receptors and enzymes and increase tissue permeability. Lipid peroxidation can result in the formation of reactive aldehydes such as acrolein and 4-hydroxy-2-nonenal (4-HNE), which are highly diffusible and are able to induce various cellular events, such as proliferation, apoptosis, and activation of signalling pathways.Citation33,Citation34 Increased levels of proteins modified by 4-HNE have been observed in airway and alveolar epithelial cells, endothelial cells, and neutrophils in smokers with airway obstruction compared with individuals without airway obstruction, suggesting a role for 4-HNE in the pathogenesis of COPD.Citation35 In addition, these reactive aldehydes can form adducts with both intracellular proteins, such as histone deacetylase (HDAC)-2, and extracellular proteins, such as collagen and fibronectin, altering their function that in turn can then impact on cell function.Citation36,Citation37 Protein nitration by reactive peroxynitrite anions (ONOO−) can also affect enzyme activity by reducing HDAC activity.Citation37,Citation38 This could explain why corticosteroids are not effective in resolving the inflammatory response in COPD patients, given that they have a much reduced HDAC-2 activity correlating with disease severity.Citation39,Citation40 Levels of nitrotyrosine formation are elevated in COPD and levels of protein nitration have been found in induced sputum cells of COPD patients.Citation41 ONOO− is also capable of attacking sulphydryl groups forming nitrosothiols, a process called nitrosylation, which can also impact upon protein function.Citation16
Endogenous antioxidant defences within the lung
In order to combat and neutralize the deleterious effects of ROS, the normal lung has various endogenous antioxidant strategies, which employ both enzymatic and non-enzymatic mechanisms. Within the lung lining fluid, several non-enzymatic antioxidant species exist, which include glutathione (GSH), vitamin C, uric acid, vitamin E, and albumin.Citation12,Citation16 GSH is present in greater concentrations in the epithelial lining fluid compared with plasma and plays an important protective role in the airspaces and epithelial cells against oxidants in the extracellular milieu.Citation42 Human studies have shown elevated levels of GSH in epithelial lining fluid in chronic cigarette smokers compared to non-smokers.Citation42,Citation43 Enzymatic antioxidant defences include SOD, catalase, thioredoxin, Gpx, and glutathione-S-transferase. Bronchial epithelial cells of rats exposed to cigarette smoke have shown increased expression of the antioxidant genes manganese superoxide dismutase, metallothionein, and Gpx.Citation44 Increased activity of SOD in alveolar macrophages from young smokers also has been reported.Citation45 However, Kondo and co-workers found that the increased superoxide generation by alveolar macrophages in elderly smokers was associated with decreased antioxidant enzyme activities when compared to non-smokers.Citation46 In addition, this reduced activity was not associated with decreased gene expression, but was due to modification at the post-translational level.Citation46 The activities of SOD and Gpx have been shown to be higher in the lungs of rats exposed to cigarette smoke.Citation47 The mechanism(s) for the induction of antioxidant enzymes in erythrocytes,Citation48 alveolar macrophages,Citation45 and lungsCitation47 by cigarette smoke exposure are currently unknown.
Glutathione peroxidases
GPxs are a family of selenium-dependent and -independent antioxidant enzymes that catalyze the reduction of damaging H2O2 as well as a large variety of hydroperoxides (such as DNA peroxides and lipid peroxides) into water and alcohols, respectively, typically using GSH as reductant and thus protect biomembranes and cellular components against oxidative stress.Citation49 According to phylogeny, the Gpx family consists of three evolutionary groups arising from a Cys-containing ancestor: Gpx-1/Gpx-2, Gpx-3/Gpx-5/Gpx-6, and Gpx-4/Gpx-7/Gpx-8, the expression levels of each isoform varying depending on the tissue type.Citation50 Despite the well known ability of Gpxs to remove H2O2, the exact role of these enzymes under physiological and oxidative stress conditions is still not clearly defined. Recent studies using knockout mice have provided data about the function of the most abundant glutathione peroxidase, Gpx-1. Mice deficient in Gpx-1 are healthy and fertile and do not show any histopathologies (organs examined included lung, liver, brain, heart, and kidney) up to 15 months of age,Citation51 consistent with a limited role for Gpx-1 during normal development and under physiological conditions.Citation52,Citation53 In addition, these studies showed that Gpx-1 plays a protective role in the cell in situations of oxidative stress.Citation51 This hypothesis is confirmed by our study showing that Gpx-1 knockout mice are susceptible to oxidative stressors such as cigarette smoke.Citation54 Moreover, Gpx-1 has been implicated in the development and prevention of many common and complex diseases, including cancer and cardiovascular disease.Citation55
Gpxs in normal physiology and lung diseases
While it is clear that there are various isoforms of Gpx, the lung literature has essentially divided them into two groups, cellular (classical) and extracellular. The alveolar epithelial lining fluid contains a very high amount of both extracellular Gpx and classical Gpx and these enzymes each contribute about half of the Gpx activity in epithelial lining fluid.Citation42 Primary bronchial epithelial cells, alveolar macrophages and other lung cell lines can synthesize classical Gpx and extracellular Gpx and also secrete the extracellular enzyme.Citation56 Classical Gpx is induced by hyperoxiaCitation57 and by the combination of hyperoxia and TNF.Citation58 The levels of both classical Gpx and extracellular Gpx are decreased after exposure to ozone.Citation59 Extracellular Gpx has been shown to protect alveolar epithelial cells against hyperoxia-induced injury in rats.Citation60 It has been shown that Gpx-2 is induced in the lungs of mice in response to cigarette smoke and that basal and CS-inducible expression of Gpx-2 are directly dependent on Nrf2.Citation61,Citation62 In humans, Gpx-1 has been shown to be increased in COPD,Citation63 and Gpx-2 showed a three- to five-fold up-regulation in epithelial cells of smokers compared with nonsmokers.Citation64–Citation66 Gpx-3 showed a two-fold up-regulation in epithelial cells of smokers compared with nonsmokers.Citation65,Citation67 There was little evidence of differential regulation of Gpx-4, Gpx-5, or Gpx-7 by disease status.Citation65 Gpx activity is significantly reduced in smokers and subjects with COPD, highlighting its prominent role in lung antioxidant defense.Citation63 With respect to reduced Gpx activity in COPD patients and smokers, erythrocyte Gpx activity was significantly lower in patients with severe COPD compared with patients with moderate COPD.Citation68 In addition, Gpx activity was decreased in plasma from COPD patientsCitation69 and total blood from smokers and ex-smokers.Citation70 It has been shown that COPD patients are deficient in selenium and that this could explain the observed reduction in Gpx activity.Citation70 Moreover, selenium is an important element in the Gpx catalysis of the reaction between GSH and ROS. There is a direct relationship between systemic Gpx activity and FEV1,Citation68 and oxidative stress correlates with both lung function and body mass index in COPD.Citation69 Elevated levels of H2O2 are measured in the exhaled breath condensate of COPD patients, particularly during exacerbations.Citation25
Therapeutic intervention with antioxidants
Given the role of oxidative stress in COPD, various approaches have been used to study the benefit of antioxidants in COPD. The strategies employed have included (1) increasing the endogenous antioxidant enzyme defences or (2) enhancing the non-enzymatic defences through dietary or pharmacological means.Citation12,Citation16 Of these strategies, enzyme mimetics have shown the most promise. Enzyme mimetics are generally small compounds that posses catalytic activity that mimics the activity of larger enzyme-based molecules. A number of SOD mimetics based around organomanganese complexes have been developed, which retain their antioxidant properties in vivo. Within the various classes of SOD mimetics only the metalloporphyrin-based compounds AEOL10150 and AEOL10113 have been studied in models of airway inflammation. In one study, AEOL10150 was demonstrated to inhibit cigarette smoke-induced lung inflammation,Citation71 suggestive of a potential therapeutic benefit in COPD. Another type of catalytic antioxidant is the Gpx mimetic ebselen. This is a selenium-based organic complex and has been shown to be a very powerful antioxidant against damaging H2O2 and the potentially destructive peroxynitrite molecule.Citation72 It is able to prevent both NFκB/AP-1 activation and pro-inflammatory gene expression in human leukocytes exposed to peroxynitrite. Other studies have shown that Ebselen is also active in vivo in ameliorating airway inflammation induced by LPS,Citation73,Citation74 ozone,Citation75 sephadex,Citation76 and influenza A virus.Citation77
Gpx-1 protects against cigarette smoke-induced lung inflammation and damage
We previously have shown that mice lacking the Gpx-1 gene are highly susceptible to oxidative stress but do not display an overt phenotype and thus proposed that Gpx-1 may be an attractive target for increasing the antioxidant capacity in ischemia/reperfusion brain injury where oxidative stress is involved.Citation51,Citation78,Citation79 Given that the known biology of Gpx-1 appears to be protection during oxidative stress, and evidence of a role in COPD, we proposed that Gpx-1 protects against cigarette smoke-induced lung inflammation and damage. Indeed, we have shown that Gpx-1-deficient mice exposed to short-term cigarette smoke had significantly elevated levels in BALF macrophages, neutrophils, proteolytic burden, and whole lung macrophage and neutrophil chemotactic factor gene expression.Citation54 Moreover, BALF from cigarette smoke-exposed Gpx-1 deficient mice had increased protease expression (MMP-9) and activity compared to wild-type cigarette smoke-exposed mice. Thus, our data provide new evidence suggesting that Gpx-1 is required to control cigarette smoke-induced lung inflammation and the potential therapeutic utility of targeting Gpx-1 in vivo. In addition, the fact that Gpx-1-deficient mice have normal BALF cell counts at baseline (i.e. no cigarette smoke exposure) strengthens the hypothesis that Gpx-1 may be protective in cigarette smoke-induced lung inflammation where there is an enhanced oxidant burden.
In our study, we did not explore the role of Gpx-1 in the development of emphysema. We and others have proposed that short-term responses to cigarette smoke exposure may be a useful predictor of the development of emphysema, and such models may be a useful screen by which to identify therapeutic targets.Citation80–Citation85 Thus, given that Gpx-1 protects against cigarette smoke-induced lung inflammation in the present study, we would predict that Gpx-1 also protects against cigarette smoke-induced emphysema. This prediction is in accordance with work by Foronjy and colleagues showing that transgenic Gpx-1 mice exposed to cigarette smoke for 12 months were protected against the formation of emphysema and airway inflammation whereas Gpx-1 knockout mice exhibited an exaggerated emphysema phenotype.Citation86
Ebselen reduces cigarette smoke-induced lung inflammation in mice
Ebselen has been shown to be protective in vivo in disease situations hallmarked by oxidative stress such as diabetes-associated atherosclerosis and cerebral ischaemia–reperfusion injury.Citation87,Citation88 In addition, ebselen has been used in clinical trials of acute ischaemic stroke.Citation89,Citation90 Specifically, Yamaguchi et al.Citation90 explored the effects of ebselen on the outcome of acute ischaemic stroke in a multi-centre, placebo-controlled, double-blind clinical trial. They demonstrated that early treatment (i.e. patients who started ebselen within 24 h of stroke onset) with ebselen (150 mg bid) improved the outcome of acute ischaemic stroke.Citation90 Similarly, Ogawa et al.Citation89 showed in a randomized, double-blind, placebo-controlled trial of ebselen conducted in patients with complete occlusion of the middle cerebral artery that ebselen protected the brain from ischaemic damage in the acute stage. In this light, it is possible that ebselen may influence the key reactions involved in the inflammatory responses in COPD, but no studies have yet been reported on the protective role of ebselen in cigarette smoke-induced lung inflammation. We showed for the first time that ebselen, when administered prophylactically and during established inflammation, reduces cigarette smoke-induced BALF inflammation in mice.Citation54 Of interest was that the approximately two-fold increase in BALF inflammation of Gpx-1-deficient mice exposed to cigarette smoke was abolished by ebselen administration. This clearly shows that there is a link between ebselen and its direct effects on Gpx-1 in smoke-exposed mice. Moreover, this is in accord with our previous study showing that pretreatment of Gpx-1 deficient mice with ebselen restored microvascular perfusion, limited the induction and activation of MMP-9, and attenuated the increases in infarct size and vascular permeability.Citation88 It is likely that ebselen reduced cigarette smoke-induced BALF inflammation by inhibiting the gene expression of a variety of neutrophil (e.g. IL-17A) and macrophage (e.g. MIP-1α and MCP-1) chemotactic factors pertinent to lung inflammation. Suppression of GM-CSF by ebselen would also have contributed to the reduced BALF inflammation given that GM-CSF is a survival factor for neutrophils and macrophages and that GM-CSF inhibits neutrophil apoptosis.Citation91–Citation93 In addition, it could be possible that ebselen's antioxidant properties of removing H2O2, scavenging ONOO−, and enhancing pulmonary expression of both copper/zinc and manganese SODs (which can contribute to a decrease in the formation of ONOO− by lowering the concentration of available O2•−) may have also contributed to reduced inflammation. In addition, it is possible that ebselen reduced cigarette smoke-induced BALF inflammation by inducing cell death as previously described by Guerin and Gauthier.Citation94 Thus, targeting Gpx-1 with mimetics such as ebselen might exert anti-inflammatory and antioxidant effects in vivo.
Conclusion
COPD is a major health problem and a significant economic burden worldwide. It is believed that an exaggerated inflammatory response to cigarette smoke in which macrophages, neutrophils, and lymphocytes are prominent leads to oxidative stress, chronic inflammation, emphysema, small airway fibrosis, mucus hypersecretion, and progressive airflow limitation. Given the increased oxidant burden in smokers and patients with COPD, we propose that the antioxidant enzyme Gpx-1 may be a novel therapeutic target for the treatment of cigarette smoke-induced lung disease. Indeed, we and others have provided evidence that the antioxidant enzyme Gpx-1 protects the lung from cigarette smoke-induced inflammation and emphysema. In addition, we have shown that increasing the antioxidant capacity of the lungs by using a Gpx mimetic such as ebselen is beneficial in resolving inflammation induced by cigarette smoke. It is important to note that ebselen was effective when given prophylactically and, perhaps more importantly, when administered therapeutically (i.e. in established disease) as would be the case in clinical practice. Thus, the striking effect of ebselen in our model suggests that Gpx-1 may be a novel target that can be exploited therapeutically to slow or prevent cigarette smoke-induced emphysema and reduce the severity of inflammation in COPD.
References
- Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003;22:672–88.
- Shapiro SD. The macrophage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999;160(5 Pt 2):S29–32.
- Pesci A, Balbi B, Majori M, Cacciani G, Bertacco S, Alciato P, et al. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur Respir J 1998;12(2):380–6.
- Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996;153(2):530–4.
- Retamales I, Elliott WM, Meshi B, Coxson HO, Pare PD, Sciurba FC, et al. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med 2001;164:469–73.
- Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 1998;158(4):1277–85.
- Barnes PJ. Macrophages as orchestrators of COPD. J COPD 2004;1:59–70.
- Finlay GA, O'Driscoll LR, Russell KJ, D'Arcy EM, Masterson JB, FitzGerald MX, et al. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med 1997;156(1):240–7.
- Ohnishi K, Takagi M, Kurokawa Y, Satomi S, Konttinen YT. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab Invest 1998;78(9):1077–87.
- Lim S, Roche N, Oliver BG, Mattos W, Barnes PJ, Fan Chung K. Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10. Am J Respir Crit Care Med 2000;162(4 Pt 1):1355–60.
- Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2002;26(5):602–9.
- Rahman I. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim Biophys Acta 2012;1822(5):714–28.
- Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 1985;64:111–26.
- Nakayama T, Church DF, Pryor WA. Quantitative analysis of the hydrogen peroxide formed in aqueous cigarette tar extracts. Free Radic Biol Med 1989;7(1):9–15.
- Pryor WA, Stone K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann N Y Acad Sci 1993;686:12–27.
- Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J 2006;28(1):219–42.
- Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med 1996;21(5):669–81.
- Drath DB, Karnovsky ML, Huber GL. The effects of experimental exposure to tobacco smoke on the oxidative metabolism of alveolar macrophages. J Reticuloendothel Soc 1979;25(6):597–604.
- Schaberg T, Klein U, Rau M, Eller J, Lode H. Subpopulations of alveolar macrophages in smokers and nonsmokers: relation to the expression of CD11/CD18 molecules and superoxide anion production. Am J Respir Crit Care Med 1995;151(5):1551–8.
- Mateos F, Brock JH, Perez-Arellano JL. Iron metabolism in the lower respiratory tract. Thorax 1998;53(7):594–600.
- Lapenna D, de Gioia S, Mezzetti A, Ciofani G, Consoli A, Marzio L, et al. Cigarette smoke, ferritin, and lipid peroxidation. Am J Respir Crit Care Med 1995;151(2 Pt 1):431–5.
- Thompson AB, Bohling T, Heires A, Linder J, Rennard SI. Lower respiratory tract iron burden is increased in association with cigarette smoking. J Lab Clin Med 1991;117(6):493–9.
- Wesselius LJ, Nelson ME, Skikne BS. Increased release of ferritin and iron by iron-loaded alveolar macrophages in cigarette smokers. Am J Respir Crit Care Med 1994;150(3):690–5.
- Nowak D, Antczak A, Krol M, Pietras T, Shariati B, Bialasiewicz P, et al. Increased content of hydrogen peroxide in the expired breath of cigarette smokers. Eur Respir J 1996;9(4):652–7.
- Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders PL, van Herwaarden CL, et al. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1996;154(3 Pt 1):813–6.
- Nakajima Y, Aoshiba K, Yasui S, Nagai A. H2O2 induces apoptosis in bovine tracheal epithelial cells in vitro. Life Sci 1999;64(26):2489–96.
- Aoshiba K, Tamaoki J, Nagai A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2001;281(6):L1392–401.
- Hoshino Y, Mio T, Nagai S, Miki H, Ito I, Izumi T. Cytotoxic effects of cigarette smoke extract on an alveolar type II cell-derived cell line. Am J Physiol Lung Cell Mol Physiol 2001;281(2):L509–16.
- Vayssier M, Banzet N, Francois D, Bellmann K, Polla BS. Tobacco smoke induces both apoptosis and necrosis in mammalian cells: differential effects of HSP70. Am J Physiol 1998;275(4 Pt 1):L771–9.
- Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 1996;154(4 Pt 1):1055–60.
- Postma DS, Renkema TE, Noordhoek JA, Faber H, Sluiter HJ, Kauffman H. Association between nonspecific bronchial hyperreactivity and superoxide anion production by polymorphonuclear leukocytes in chronic air-flow obstruction. Am Rev Respir Dis 1988;137(1):57–61.
- Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 1995;41(12 Pt 2):1819–28.
- Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem 1999;274(4):2234–42.
- Parola M, Bellomo G, Robino G, Barrera G, Dianzani MU. 4-Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid Redox Signal 1999;1(3):255–84.
- Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, et al. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002;166(4):490–5.
- Kirkham PA, Spooner G, Rahman I, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem Biophys Res Commun 2004;318(1):32–7.
- Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, et al. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol 2004;31(6):633–42.
- Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun 2004;315(1):240–5.
- Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med 2004;200(5):689–95.
- Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med 2005;352(19):1967–76.
- Ichinose M, Sugiura H, Yamagata S, Koarai A, Shirato K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am J Respir Crit Care Med 2000;162(2 Pt 1):701–6.
- Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol 1987;63(1):152–7.
- Morrison D, Rahman I, Lannan S, MacNee W. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am J Respir Crit Care Med 1999;159(2):473–9.
- Gilks CB, Price K, Wright JL, Churg A. Antioxidant gene expression in rat lung after exposure to cigarette smoke. Am J Pathol 1998;152(1):269–78.
- McCusker K, Hoidal J. Selective increase of antioxidant enzyme activity in the alveolar macrophages from cigarette smokers and smoke-exposed hamsters. Am Rev Respir Dis 1990;141(3):678–82.
- Kondo T, Tagami S, Yoshioka A, Nishimura M, Kawakami Y. Current smoking of elderly men reduces antioxidants in alveolar macrophages. Am J Respir Crit Care Med 1994;149(1):178–82.
- York GK, Peirce TH, Schwartz LW, Cross CE. Stimulation by cigarette smoke of glutathione peroxidase system enzyme activities in rat lung. Arch Environ Health 1976;31(6):286–90.
- Toth KM, Berger EM, Beehler CJ, Repine JE. Erythrocytes from cigarette smokers contain more glutathione and catalase and protect endothelial cells from hydrogen peroxide better than do erythrocytes from nonsmokers. Am Rev Respir Dis 1986;134(2):281–4.
- Brigelius-Flohe R. Glutathione peroxidases and redox-regulated transcription factors. Biol Chem 2006;387(10–11):1329–35.
- Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta 2013;1830(5):3289–303.
- de Haan JB, Bladier C, Griffiths P, Kelner M, O'Shea RD, Cheung NS, et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J Biol Chem 1998;273(35):22528–36.
- Ho YS, Magnenat JL, Bronson RT, Cao J, Gargano M, Sugawara M, et al. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem 1997;272(26):16644–51.
- Cheng WH, Ho YS, Ross DA, Valentine BA, Combs GF, Lei XG. Cellular glutathione peroxidase knockout mice express normal levels of selenium-dependent plasma and phospholipid hydroperoxide glutathione peroxidases in various tissues. J Nutr 1997;127(8):1445–50.
- . Duong C, Seow HJ, Bozinovski S, Crack P, Anderson GP, Vlahos R. Glutathione peroxidase-1 protects against cigarette smoke-induced lung inflammation in mice. Am J Physiol Lung Cell Mol Physiol 2010;299(3):L425–33.
- Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 2011;15(7):1957–97.
- Avissar N, Finkelstein JN, Horowitz S, Willey JC, Coy E, Frampton MW, et al. Extracellular glutathione peroxidase in human lung epithelial lining fluid and in lung cells. Am J Physiol 1996;270(2 Pt 1):L173–82.
- Jornot L, Junod AF. Response of human endothelial cell antioxidant enzymes to hyperoxia. Am J Respir Cell Mol Biol 1992;6(1):107–15.
- Tsan MF, White JE, Santana TA, Lee CY. Tracheal insufflation of tumor necrosis factor protects rats against oxygen toxicity. J Appl Physiol 1990;68(3):1211–9.
- Avissar NE, Reed CK, Cox C, Frampton MW, Finkelstein JN. Ozone, but not nitrogen dioxide, exposure decreases glutathione peroxidases in epithelial lining fluid of human lung. Am J Respir Crit Care Med 2000;162(4 Pt 1):1342–7.
- Roum JH, Aledia AS, Carungcong LA, Kim KJ, Borok Z. Extracellular glutathione inhibits oxygen-induced permeability changes in alveolar epithelial monolayers. J Appl Physiol 2001;91(2):748–54.
- Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 2004;114(9):1248–59.
- Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohe R, et al. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol 2006;35(6):639–50.
- Bentley AR, Emrani P, Cassano PA. Genetic variation and gene expression in antioxidant related enzymes and risk of COPD: a systematic review. Thorax 2008;63(11):956–61.
- Hackett NR, Heguy A, Harvey BG, O'Connor TP, Luettich K, Flieder DB, et al. Variability of antioxidant-related gene expression in the airway epithelium of cigarette smokers. Am J Respir Cell Mol Biol 2003;29(3 Pt 1):331–43.
- Pierrou S, Broberg P, O'Donnell RA, Pawlowski K, Virtala R, Lindqvist E, et al. Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007;175(6):577–86.
- Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, et al. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci USA 2004;101(27):10143–8.
- Comhair SA, Thomassen MJ, Erzurum SC. Differential induction of extracellular glutathione peroxidase and nitric oxide synthase 2 in airways of healthy individuals exposed to 100% O(2) or cigarette smoke. Am J Respir Cell Mol Biol 2000;23(3):350–4.
- Kluchova Z, Petrasova D, Joppa P, Dorkova Z, Tkacova R. The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol Res 2007;56(1):51–6.
- Vibhuti A, Arif E, Deepak D, Singh B, Qadar Pasha MA. Correlation of oxidative status with BMI and lung function in COPD. Clin Biochem 2007;40(13–14):958–63.
- Santos MC, Oliveira AL, Viegas-Crespo AM, Vicente L, Barreiros A, Monteiro P, et al. Systemic markers of the redox balance in chronic obstructive pulmonary disease. Biomarkers 2004;9(6):461–9.
- Smith KR, Uyeminami DL, Kodavanti UP, Crapo JD, Chang LY, Pinkerton KE. Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Radic Biol Med 2002;33(8):1106–14.
- Jozsef L, Filep JG. Selenium-containing compounds attenuate peroxynitrite-mediated NF-kappaB and AP-1 activation and interleukin-8 gene and protein expression in human leukocytes. Free Radic Biol Med 2003;35(9):1018–27.
- Haddad el B, McCluskie K, Birrell MA, Dabrowski D, Pecoraro M, Underwood S, et al. Differential effects of ebselen on neutrophil recruitment, chemokine, and inflammatory mediator expression in a rat model of lipopolysaccharide-induced pulmonary inflammation. J Immunol 2002;169(2):974–82.
- Zhang M, Nomura A, Uchida Y, Iijima H, Sakamoto T, Iishii Y, et al. Ebselen suppresses late airway responses and airway inflammation in guinea pigs. Free Radic Biol Med 2002;32(5):454–64.
- Ishii Y, Hashimoto K, Hirano K, Morishima Y, Mochizuki M, Masuyama K, et al. Ebselen decreases ozone-induced pulmonary inflammation in rats. Lung 2000;178(4):225–34.
- Belvisi MG, Haddad EB, Battram C, Birrell M, Foster M, Webber S. Anti-inflammatory properties of ebselen in a model of sephadex-induced lung inflammation. Eur Respir J 2000;15(3):579–81.
- Yatmaz S, Seow HJ, Gualano RC, Wong ZX, Stambas J, Selemidis S, et al. Glutathione peroxidase-1 reduces influenza A virus-induced lung inflammation. Am J Respir Cell Mol Biol 2013;48(1):17–26.
- Crack PJ, Taylor JM, Flentjar NJ, de Haan J, Hertzog P, Iannello RC, et al. Increased infarct size and exacerbated apoptosis in the glutathione peroxidase-1 (Gpx-1) knockout mouse brain in response to ischemia/reperfusion injury. J Neurochem 2001;78(6):1389–99.
- Knorpp T, Robinson SR, Crack PJ, Dringen R. Glutathione peroxidase-1 contributes to the protection of glutamine synthetase in astrocytes during oxidative stress. J Neural Transm 2006;113(9):1145–55.
- Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med 2002;166(6):849–54.
- Churg A, Zay K, Shay S, Xie C, Shapiro SD, Hendricks R, et al. Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am J Respir Cell Mol Biol 2002;27(3):368–74.
- Vlahos R, Bozinovski S, Gualano RC, Ernst M, Anderson GP. Modelling COPD in mice. Pulm Pharmacol Ther 2006;19(1):12–7.
- Vlahos R, Bozinovski S, Jones JE, Powell J, Gras J, Lilja A, et al. Differential protease, innate immunity, and NF-kappaB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol 2006;290(5):L931–45.
- Stevenson CS, Birrell MA. Moving towards a new generation of animal models for asthma and COPD with improved clinical relevance. Pharmacol Ther 2011;130(2):93–105.
- Wright JL, Churg A. Animal models of cigarette smoke-induced chronic obstructive pulmonary disease. Expert Rev Respir Med 2010;4(6):723–34.
- Geraghty P, Hardigan A, Wallace AM, Mirochnitchenko O, Thankachen J, Arellanos L, et al. The GPx1-PTP1B-PP2A axis: a key determinant of airway inflammation and alveolar destruction. Am J Respir Cell Mol Biol 2013; Apr 5 [Epub ahead of print].
- Chew P, Yuen DY, Koh P, Stefanovic N, Febbraio MA, Kola I, et al. Site-specific antiatherogenic effect of the antioxidant ebselen in the diabetic apolipoprotein E-deficient mouse. Arterioscler Thromb Vasc Biol 2009;29(6):823–30.
- Wong CH, Bozinovski S, Hertzog PJ, Hickey MJ, Crack PJ. Absence of glutathione peroxidase-1 exacerbates cerebral ischemia-reperfusion injury by reducing post-ischemic microvascular perfusion. J Neurochem 2008;107(1):241–52.
- Ogawa A, Yoshimoto T, Kikuchi H, Sano K, Saito I, Yamaguchi T, et al. Ebselen in acute middle cerebral artery occlusion: a placebo-controlled, double-blind clinical trial. Cerebrovasc Dis 1999;9(2):112–8.
- Yamaguchi T, Sano K, Takakura K, Saito I, Shinohara Y, Asano T, et al. Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke 1998;29(1):12–7.
- Hamilton JA. GM-CSF in inflammation and autoimmunity. Trends Immunol 2002;23(8):403–8.
- Vlahos R, Bozinovski S, Chan SP, Ivanov S, Linden A, Hamilton JA, et al. Neutralizing granulocyte/macrophage colony-stimulating factor inhibits cigarette smoke-induced lung inflammation. Am J Respir Crit Care Med 2010;182(1):34–40.
- Vlahos R, Bozinovski S, Hamilton JA, Anderson GP. Therapeutic potential of treating chronic obstructive pulmonary disease (COPD) by neutralising granulocyte macrophage-colony stimulating factor (GM-CSF). Pharmacol Ther 2006;12(1):106–15.
- Guerin PJ, Gauthier ER. Induction of cellular necrosis by the glutathione peroxidase mimetic ebselen. J Cell Biochem 2003;89(1):203–11.