Abstract
Mutations in mismatch repair (MMR) genes are commonly associated with the development of colorectal cancer. Additionally, base excision repair, which involves apurinic/apyrimidinic endonuclease 1 (APE1), recognizes and eliminates oxidative DNA damage. Here, we investigated the possible roles of APE1 in dextran sulfate sodium (DSS)-induced acute colitis using the young rat model. Four-week-old Sprague–Dawley rats were administered 2% DSS in drinking water for 1 week. MMR and APE1 expression levels were assessed by western blotting and immunohistochemistry. Following DSS treatment, growth of young rats failed and the animals had loose stools. Together with the histological changes associated with acute colitis, APE1 and MSH2 levels increased significantly at 3 and 5 days after DSS treatment, respectively. The difference between APE1 and MSH2 expression was significant. DSS-induced DNA damage and subsequent repair activity were evaluated by staining for 8-hydroxy-deoxyguanosine (8-OHdG) and APE1, respectively; 8-OHdG immunoreactivity increased throughout the colonic mucosa, while APE1 levels in the surface epithelium increased at an earlier timepoint. Taken together, our data suggest that changes in APE1 expression after DSS treatment occurred earlier and were more widespread than changes in MMR expression, suggesting that APE1 is more sensitive for prediction of DNA deterioration in DSS-induced colitis.
Introduction
The term ‘inflammatory bowel disease (IBD)’ applies to a group of idiopathic chronic inflammatory disorders involving the gastrointestinal tract, and includes ulcerative colitis (UC) and Crohn's disease. IBD is more common in Caucasians, and patients with chronic IBD have an increased incidence of carcinoma compared with the general population. Although it is less common in non-Caucasian populations, the prevalence of IBD is higher in Koreans.Citation1 Experimental IBD models are essential for investigation of the pathogenesis of this group of conditions due to limitations regarding patients and clinical trials in the Republic of Korea. Several agents, including dextran sulfate sodium (DSS), have been used to induce experimental inflammation for the study of IBDs.Citation2,Citation3 DSS-induced colitis in adult rodents results in inflammation in mainly the distal colonic mucosa with significant mucosal infiltration of neutrophils. DSS-induced colitis is characterized by an initial acute colonic injury, which leads to chronic colitis upon oral administration of several alternating cycles of DSS and water; the pathology resembles that of UC.Citation3–Citation6
The information available regarding the molecular and genetic pathways that result in colorectal cancer, such as tumor suppressor genes, DNA mismatch repair (MMR) genes and proto-oncogenes, has increased markedly. The specific contributing mutations in genes such as adenomatous polyposis coli (APC)Citation7–Citation11 and MMRCitation8,Citation12–Citation18 have been investigated intensively. Mutations or deficiencies in APC and MMR contribute significantly to the development of age-related or familial colonic neoplasia and sporadic or mucosal damage-related neoplasia, respectively. Additionally, base excision repair (BER) factors, including Ogg1, MutY DNA glycosylase, and MutY homolog (MYH), recognize and excise oxidative DNA damage in the colon.Citation19–Citation21 BER activities are associated with increased oxidative DNA damage, including 8-oxoguanine and 8-hydroxy-deoxyguanosine (8-OHdG).Citation19,Citation21 Specifically, mutations in the MYH gene are responsible for colorectal cancer, and the activity of the encoded protein is modulated by several factors, including proliferating cell nuclear antigen (PCNA), MMR enzymes (MSH2/MSH6), and apurinic/apyrimidinic endonuclease 1 (APE1).Citation20,Citation21
Colitis can be induced in young rodents by lower DSS concentrations (1–2% w/v) compared to adults (5% w/v) because young animals show increased sensitivity to DSS.Citation22 The syndrome evoked in young rats by a lower DSS concentration is similar to that in adult animals, although in young animals, symptoms appear earlier and toxicity is greater than in adults. In this model, the roles of tumor suppressor genes or oncogenes associated with aging may be excluded, and DNA repair pathways related to mucosal damage are active. BER has been reported during colitis-related cancer development. However, the roles of APE1 (also known as Redox factor 1 (Ref-1)) in this pathway are unclear, although it is known that MYH can be modulated by APE1.Citation20 Previous studies have suggested that APE1 deficiency increases the occurrence of DNA abasic (apurinic/apyrimidinic (AP0) sites, resulting in slow BER in cultured colon cancer cells;Citation23–Citation25 indeed, APE1 expression is increased in intestinal adenomas.Citation26 Therefore, we hypothesized a close relationship between APE1 and the development of DSS-induced acute colitis. We thus investigated the correlation between the APE1 and MMR responses in young rats and identified the cells that express APE1 in response to DSS-induced colitis.
Methods
Animals and treatments
The experiments herein were performed using 4-week-old Sprague–Dawley rats (Da-mool Science, Daejeon, Republic of Korea, 57.9 ± 4.5 g). The animals were housed under standard conditions in a controlled environment with a 12-hour dark–light cycle and access to food and water ad libitum. All experimental procedures and care of animals were conducted in accordance with the guidelines of Chosun University's Animal Care and Use Committee.
The DSS-induced colitis model was as described previously.Citation22 Animals were randomly assigned to five groups. Four groups (n = 7/group) received DSS (MW 40 kDa, 2% w/v, ICN Biomedicals Aurora, OH) in the drinking water, which was prepared fresh daily. Untreated control animals (n = 7) received tap water only. Changes in growth rate compared to the control group, stool consistency, and the presence of gross bleeding in feces were checked throughout the study period.
Animals were sacrificed 1 (DSS-1d), 3 (DSS-3d), 5 (DSS-5d), or 7 (DSS-7d) days after DSS treatment; rats without DSS treatment (DSS-0d) served as controls. Whole colons were removed by excising at positions 2 cm distal from the cecum and rectum, and western blotting and immunohistochemistry were performed in accordance with an established method.
Antibodies
Primary antibodies used were: monoclonal anti-APC (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), monoclonal anti-APE1 (Santa Cruz Biotech-nology Inc.), polyclonal anti-MSH2 (BioVision Research Products, Mountain View, CA, USA), monoclonal anti-MSH6 (BD Biosciences Pharmingen, San Diego, CA, USA), monoclonal anti-8-hydroxy-2′-deoxyguanosine (8-OHdG, JaICA, Shizuoka, Japan), and polyclonal anti-β-actin (Santa Cruz Biotechnology Inc.).
Western blotting
Pooled isolated colon sections from each group (n = 4/group) were suspended in 1 ml of cold homogenizing buffer (20 mM HEPES, pH 7.4, 2 mM EGTA, 50 mM glycerol phosphate, 1% Triton X-100, 10% glycerol, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 10 g/ml leupeptin, 10 g/ml aprotinin, 1 mM NasVO4, and 5 mM NaF) containing a protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), and homogenized three times using an ultrasonic cell disruptor (Branson Ultrasonics Co., Danbury, CT) for 30 seconds each at 30-second intervals, followed by centrifugation at 10 000 × g for 10 minutes at 4°C. Supernatant protein concentrations were determined using a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, USA). An aliquot of the supernatant (30-μg protein) was then suspended in 20-μl loading buffer composed of a 1:1 mixture (v/v) of the homogenizing buffer mentioned above and sample buffer (50 mM Tris-HCl, pH 6.5, 0.5 mg/ml bromophenol blue, 10% glycerol, 10% sodium dodecyl sulfate, and 1% β-mercaptoethanol). The mixture was then boiled for 5 minutes, subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and transferred to polyvinyldifluoridine membranes (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA).
Immunoblotting was performed using each primary antibody. Horseradish peroxidase-linked secondary antibodies (GE Healthcare Bio-Sciences Corp.) were diluted 1:4000. The blotted proteins were then detected using the iNtRON Biotech Enhanced Chemiluminescence Detect System (Seoul, Republic of Korea) and quantified using ImageQuant 350 (GE Healthcare Korea, Seoul, Republic of Korea). Data are expressed as densitometric units of each primary antibody relative to β-actin, and in reference to the value of the control sample for each gel.
Histology and immunohistochemistry
After fixation with 4% paraformaldehyde, colons (n = 3/group) were embedded in paraffin wax (Tissue-Tek, Sakura, Japan) using standard procedures. Next, 5-mm-thick serial sections were cut using a Leica RM 2155 rotary microtome (Nussloch, Germany) and mounted onto slides coated with 3-aminopropyl-tri-ethoxy-silane (Sigma-Aldrich, St Louis, MO, USA). Randomly selected samples were stained with Harris’ hematoxylin and eosin (Sigma-Aldrich) for 4 minutes and 20 seconds, respectively, using a routine protocol.
Immunohistochemical staining was carried out by the routine method. In brief, incubation with primary antibodies was performed for 24 hours at 4°C. Binding was visualized using an ImmPRESS™ avidin–biotin–peroxidase kit (Vector Laboratories Inc., Burlingame, CA, USA) according to the manufacturer's instructions. Omission of incubation with the primary or secondary antibody served as a control for false positives. Immunolabeled images were captured directly using a digital camera (PL-B873CU, PixeLINK, Ottawa, Canada) and Olympus BX-51 microscope (Olympus Corp., Tokyo, Japan). Captured images were saved and subsequently processed using Adobe Photoshop (Adobe Systems, San Jose, CA, USA). The brightness and contrast of the images were adjusted only for the purpose of background consistency. Immunohistochemical staining results were interpreted and scored as follows: 0 (negative), 1+ (focal, weak staining), 2+ (diffuse, weak- or focal, strong staining), and 3+ (diffuse, strong staining).
Statistical analysis
Growth rates were assessed by comparing the weights of rats according to treatment status expressed as mean ± standard deviation (SD) using Student's t-test. Densitometry results are expressed as mean ± standard error (SE) of three independent determinations. The statistical significance of differences in protein expression according to time was assessed by one-way analysis of variance (ANOVA). Two-way ANOVA was used to compare protein expression according to protein class after adjusting for time. All statistical tests were two-tailed, with a significance level of P = 0.05. All analyses were performed using the SPSS software, version 12.0 (SPSS Inc., Chicago, IL, USA).
Results
Biological effects of DSS treatment
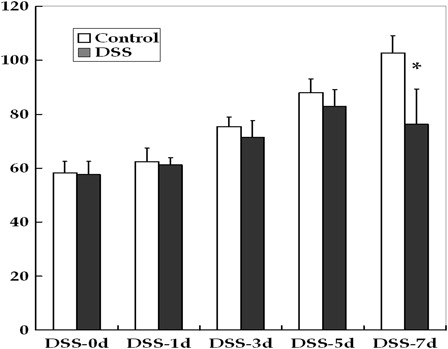
Oral administration of DSS induced acute colitis in young rats. The mortality rate reached 30% by DSS-7d; dead rats were excluded from further analyses. Rats treated with DSS exhibited symptoms including growth failure, loose stools and bloody diarrhea. At DSS-5d, 70% of the animals had loose stools, and almost all animals had bloody diarrhea at DSS-7d. There were no significant differences in the weights of rats until DSS-5d (). At DSS-7d, the weight of the DSS-treated group was significantly reduced compared to the control (P < 0.01).
Figure 1. Changes in the body weights of young rats in the control and DSS-induced colitis groups. DSS treatment caused growth failure at the end of the study period (*P < 0.01).
Histological changes after DSS treatment
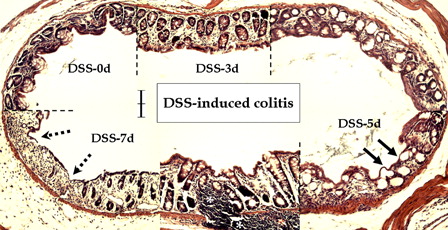
Compared to the normal colonic mucosa, inflammatory reactions involving primarily the mucosa were observed after DSS treatment (). Early histological changes at DSS-3d included vascular congestion (data not shown), focal superficial erosion and compensatory hyperplasia. Surface mucosal cells, as well as the crypt epithelium, were involved in the inflammatory reaction, which progressed to surface epithelial damage – flat dysplasia (arrows) – at DSS-5d. Dysplastic lesions, including deep ulceration of the mucosa (dotted arrows), and increased inflammatory cell infiltration of the lamina propria (asterisks), were observed at DSS-7d. Additionally, loss of goblet cells and neighboring reactive hyperplasia were observed.
Figure 2. Microscopic features of DSS-induced colitis in young rats. Compared to the control (DSS-0d), DSS treatment caused degenerative changes in the mucosal layer in a time-dependent manner. Note compensatory hyperplasia on days 3 (DSS-3d) and 7 (DSS-7d), flat dysplasia (arrows) on day 5 (DSS-5d), and ulcerative changes (dotted arrows) and infiltration of inflammatory cells in the lamina propria (asterisks) at day 7 after DSS treatment. Scale bar = 100 µm.
Western blotting after DSS treatment
MSH2 and APE1 expression levels were determined by western blotting and densitometry, and normalized to that of β-actin (). MSH2 expression levels were 1.00 ± 0.00 at DSS-0d, 0.80 ± 0.15 at DSS-1d, 0.83 ± 0.15 at DSS-3d, 2.30 ± 0.32 at DSS-5d, and 2.07 ± 0.17 at DSS-7d. Only the difference in MSH2 expression between DSS-3d and DSS-5d was significant (P < 0.05). APE1 expression levels were 1.00 ± 0.00 at DSS-0d, 1.47 ± 0.22 at DSS-1d, 3.03 ± 0.43 at DSS-3d, 4.47 ± 0.52 at DSS-5d, and 3.37 ± 0.17 at DSS-7d. Only the difference in APE1 expression at DSS-1d and DSS-3d was significant (P < 0.05).
Figure 3. Western blotting and densitometry evaluations of young rats with DSS-induced colitis. Compared to the previous time point, APE1 and MSH2 levels were significantly higher at DSS-3d and DSS-5d, respectively (*P < 0.05). A significant difference was also noted between MSH2 and APE1 at the same time point (#P < 0.05); this resulted in significantly different curves (P < 0.01).
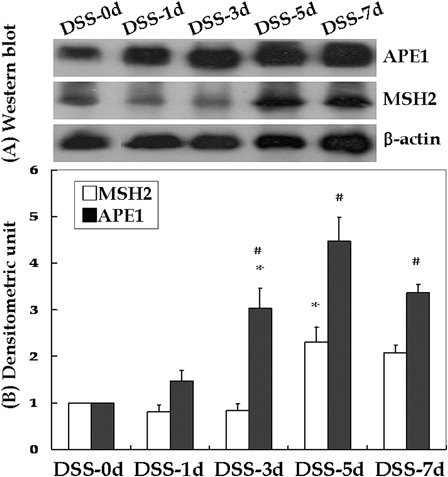
Although there were no marked differences in MSH2 and APE1 expression levels according to treatment status at DSS-0d and DSS-1d, the differences at DSS-3d (P < 0.01), DSS-5d (P < 0.05) and DSS-7d (P < 0.01) were significant. The difference in MSH2 and APE1 expression after adjusting for time was also significant (P < 0.01).
Immunohistochemical staining of APC, MSH6, 8-OHdG, and APE1 after DSS treatment
Transformation of mucosal dysplasia to tumor after DSS treatment was verified by staining for APC and MSH6, and DNA damage and its repair induced by DSS treatment were identified by 8-OHdG and APE1 staining (). According to the mucosal degeneration after DSS treatment, immunolocalization of each marker was evident in colonic mucosa free from dysplastic foci. The intensity of each marker in the surface epithelium and crypt mucosa tended to increase in a time-dependent manner. Changes in levels of the markers were evident 5 and 7 days after DSS treatment, with the exception of APE1, which showed a marked change in the surface epithelium 3 days after DSS treatment.
Table 1. The immunohistochemical staining results with APC, MSH6, 8-OHdG, and APE1 after DSS treatment
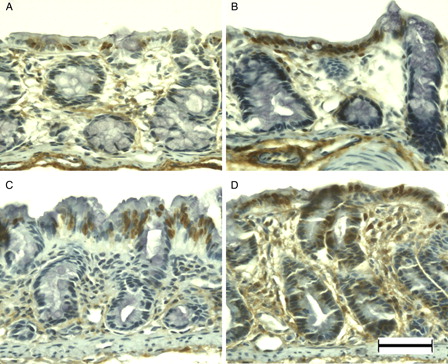
MSH6 was weakly immunopositive in primarily the surface epithelium and crypt cells in the normal colonic mucosa of young rats (A). Immunoreactivity did not change markedly at DSS-3d (B); however, at DSS-5d the majority of enterocytes were immunopositive (C). Dysplastic mucosa showed blurred and weak immunopositivity at DSS-7d (D). Although APC was observed only rarely in the normal colonic mucosa of young rats (A), immunoreactivity was evident in the nuclei of surface epithelial cells at DSS-3d (B), and enhanced at DSS-5d (C). In addition, APC-immunopositive cells were observed in the dysplastic mucosa and in cells infiltrating the lamina propria at DSS-7d (D).
Figure 4. MSH6 immunohistochemistry of DSS-induced colitis in young rats. MSH6 was immunopositive at the surface epithelium in the colon of young rats (A), and at DSS-3d (B). Compared to DSS-3d, MSH6 was immunopositive in the majority of enterocytes at DSS-5d (C), and blurred in the entire colon at DSS-7d (D). Scale bar = 50 µm.
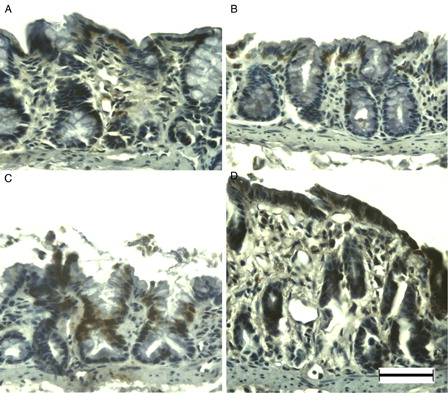
Figure 5. APC immunohistochemistry of DSS-induced colitis in young rats. APC was immunonegative in the colon of young rats (A), but was immunopositive at DSS-3d (B). APC immunoreactivity was more evident in the surface epithelium at DSS-5d (C) and in the lamina propria (asterisk) at DSS-7d (D). Scale bar = 50 µm.
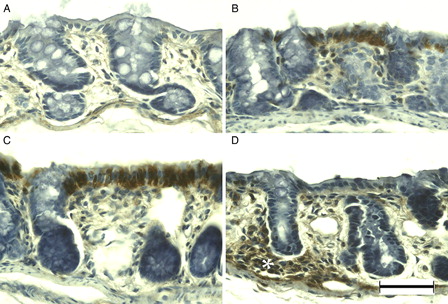
8-OHdG immunoreactivity was observed primarily in the surface epithelium and crypt cells of normal colonic mucosa (A). After DSS treatment, the number of immunopositive cells had increased in the surface epithelium (B) and crypt cells (C) at DSS-3d and DSS-5d, respectively. Almost all enterocytes were immunopositive for 8-OHdG at DSS-7d (D). APE1 immunoreactivity was evident in mainly the surface epithelium of the normal colonic mucosa of young rats (A). Immunoreactivity increased primarily in the surface epithelium at DSS-3d (B) and DSS-5d (C). APE1 was immunolocalized in the normal and slightly dysplastic mucosa at DSS-7d (D); however, the dysplastic and ulcerated areas were devoid of APE1 immunoreactivity (data not shown).
Figure 6. 8-OHdG immunohistochemistry of DSS-induced colitis in young rats. 8-OHdG was immunopositive at the surface epithelium in the colon of young rats (A). After DSS treatment, 8-OHdG immunoreactivity in the surface epithelium increased at DSS-3d (B), in crypt cells at DSS-5d (C), and in the vast majority of enterocytes at DSS-7d (D). Scale bar = 50 µm.
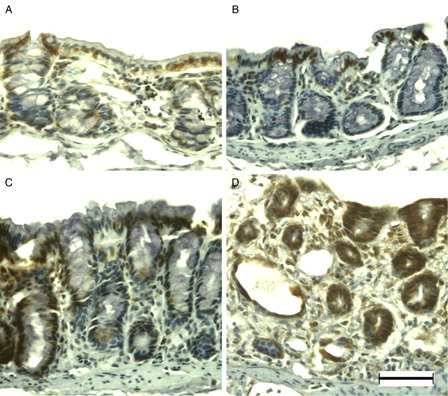
Figure 7. APE1 immunohistochemistry of DSS-induced colitis in young rats. APE1 was immunopositive at the surface epithelium in the colon of young rats (A). Immunoreactivity increased primarily in the surface epithelium at DSS-3d (B) and DSS-5d (C). APE1 was immunolocalized in the normal and slightly dysplastic mucosa at DSS-7d (D). Scale bar = 50 µm.
Discussion
Our data suggest that acute response of APE1 expression exhibited earlier and more widespread changes than did MMR in DSS-induced colitis in young rats. Growth of young rats failed and animals had loose stools after 2% DSS treatment. In addition to the histological changes associated with acute colitis, APE1 and MSH2 levels were significantly higher at DSS-3d and DSS-5d, respectively. DNA damage throughout the colonic mucosa increased with time, and APE1-mediated DNA repair activity increased in primarily the surface epithelium.
Oxidative stress has been identified as an important factor in several diseases. Two major strategies, repair and removal, have evolved to cope with oxidative-stress-mediated DNA damages. Various DNA repair pathways are activated upon oxidative DNA damage.Citation27 We hypothesized that the roles of APC associated with aging may be absent while DNA repair pathways associated with mucosal damage would be present in the DSS-induced acute colitis young rat model, because mutations or deficiencies in APC and MMR contribute significantly to the development of age-related or familial colonic neoplasia and sporadic or mucosal-damage-related neoplasia, respectively.Citation3,Citation8,Citation17 As expected, young rats failed to gain body weight and had loose stools after DSS treatment, resulting in significantly different growth curves (P < 0.01). Together with the histological changes associated with acute colitis, APC immunolocalization was evident in the surface epithelium and lamina propria at the end of the study.
Dysplastic mucosal lesions were identified by APC staining; MMR and BER levels were then determined. MMR contributes to the development of both sporadic and mucosal-damage-related neoplasia.Citation8,Citation12–Citation18 We were interested in the relationship between MMR and BER in preventing DNA damage against DSS-induced colitis in young rats because MYH, which is involved in BER, is also associated with the development of colorectal cancer, and its activity is modulated by MMR and APE1.Citation20,Citation21 We found that APE1 and MSH2 levels increased significantly at DSS-3d and DSS-5d, respectively. The difference in MSH2 and APE1 expression was significant. Time curves for MSH2 and APE1 in DSS-induced acute colitis young rat models indicated that APE1 was expressed earlier and in a more widespread manner than MSH2. Similarly, APE1 and MSH6 immunolocalization was increased primarily at DSS-3d and DSS-5d, respectively. Unlike MMR, APE levels decreased significantly between 5 and 7 days after induction of colitis. This may be because APE1 was immunopositive in primarily the surface epithelium while MSH6 was expressed in the majority of enterocytes; these reactions were associated with increased oxidative damage (8-OHdG) to the entire colonic mucosa after DSS treatment. These results suggest that mucosal cells produce MMR, but the surface epithelium does not produce APE1 when oxidative damage to the colonic mucosa is intensified by DSS administration.
The adenoma–carcinoma sequence is the process by which most colorectal carcinomas develop. APC acts during the earlier stage of dysplastic foci formation, while MMR is involved in tumor progression several years later.Citation28 In this sequence, expression of MMR in response to dysplastic changes may be belated. Therefore, an alternative DNA repair pathway is likely involved in the process of dysplastic change; we hypothesized that BER was the best candidate. While reports of the association of BER with the development of colorectal cancer are rare, with the exception of a few regarding Ogg1 and MYH,Citation19–Citation21 we hypothesized that APE1 is another important candidate. APE1 is a dual-function protein that serves as an endonuclease for abasic sites in BER and modulates or activates several transcription factors, including p53, NF-κB, Egr-1, c-Myb, HLF, and Pax-8.Citation27,Citation29,Citation30 The association between p53 and APE1 as a key regulator of apoptosis attracted our attention. APE1 acts as a regulator of p53, and vice versa. p53 downregulates APE1 expression in response to severe DNA damage, thereby promoting apoptosis of human colorectal cancer cells.Citation23–Citation25 The relationship between p53 and carcinogenesis in a DSS model has been reported.Citation31 APE1 expression, however, is increased in only approximately half of intestinal adenomas.Citation26 These findings also suggest the importance of dysregulated BER in the development of colitis-associated cancer. Immunolocalization of APE1 was more evident in the surface epithelium, but ulcerative foci were devoid of immunoreactivity. Lack of the protective surface epithelium is considered a possible cause of the acute inflammation that may be linked to growth failure, loose stools, bloody diarrhea, and eventually colon carcinogenesis. However, we cannot rule out the possibility of redox effects of APE1 in the colonic mucosa because APE1 can also regulate the transcription factors involved in DSS-induced colitis. In terms of the spectrum of pathology from inflammation to cancer, MMR immunoreactivity is a reliable marker of the transition to colitis-associated cancer. However, we anticipated that BER, which is expressed earlier than MMR, prior to the transition to cancer, would be an earlier indicator of disease progression. This notion warrants further research.
Taken together, our results demonstrate that APE1 expression changed at an earlier timepoint and was more widespread than MMR expression after DSS treatment in young rats. These results suggest that APE1 is more sensitive for prediction of DNA deterioration in DSS-induced acute colitis, as chronic mucosal dysplasia may be associated with colonic carcinogenesis. Further evidence should be collected using both other animal models and human tissues.
Acknowledgments
We thank Professor Kyeong-Soo Park (Department of Preventive Medicine, Seonam University Medical School, Republic of Korea) for statistical analysis. This research was supported by the research grant of Jeju National University in 2012.
References
- Chang DK, Kim YH, Byeon JS. The current status of ulcerative colitis-associated colorectal cancer in Korea: A KASID study. Korean J Gastroenterol 2005;46:276–82.
- Elson CO, Sartor RB, Tennyson GS, Riddel RH. Experimental models of inflammatory bowel disease. Gastroenterology 1995;109:1344–67.
- Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin 2007;28:1450–9.
- Okayasu I, Hatakeyama S, Yamada M, Okhusa T, Inagaki Y, Nakaya R. A novel method of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990;98:694–702
- Gaudio E, Taddei G, Vetushi A, Sferra R, Frieri G, Ricciardi G, et al. Dextran sulfate sodium (DSS) in rats: clinical, structural, and ultrastructural aspects. Dig Dis Sci 1999;44:1458–75.
- Cooper HS, Murthy S, Kido K, Yoshitake H, Flangan A. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-cetanin and p53 expression and the role of inflammation. Carcinogenesis 2000;21:757–68.
- Cooper HS, Everley L, Chang WC, et al. The role of mutant Apc in the development of dysplasia and cancer in the mouse model of dextran sulfate sodium-induced colitis. Gastroenterology 2001;121:1407–16.
- Narayan S, Roy D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol Cancer 2003;2:41.
- Tanaka T, Kohno H, Suzuki R, Hata K, Sugie S, Niho N, et al. Dextran sodium sulfate strongly promotes colorectal carcinogenesis in Apc (Min/+) mice: inflammatory stimuli by dextran sodium sulfate results in development of multiple colonic neoplasms. Int J Cancer 2006;118:25–34.
- Yoshimi K, Tanaka T, Takizawa A, Kato M, Hirabayashi M, Mashimo T, et al. Enhanced colitis-associated colon carcinogenesis in a novel Apc mutant rat. Cancer Sci 2009;100:2022–7.
- Alferez DG, Ryan AJ, Goodlad RA, Wright NA, Wilkinson RW. Effects of vandetanib on adenoma formation in a dextran sodium sulphate enhanced Apc(MIN/+) mouse model. Int J Oncol 2010;37:767–72.
- Fogt F, Zimmerman RL, Poremba C, Noffsinger AE, Alsaigh N, Rueschoff J. Immunohistochemical screening of mismatch repair genes hMLH1, hMSH2, and hMSH6 in dysplastic lesions of the colon. Appl Immunohistochem Mol Morphol 2002;10:57–61.
- Clark AJ, Barnetson R, Farrington SM, Dunlop MG. Prognosis in DNA mismatch repair deficient colorectal caner: are all MSI tumours equivalent? Fam Cancer 2004;3:85–91.
- Edelmann L, Edelmann W. Loss of DNA mismatch repair function and cancer predisposition in the mouse: animal models for human hereditary nonpolyposis colorectal cancer. Am J Med Genet C Semin Med Genet 2004;129C:91–9.
- Marra G, Jiricny J. DNA mismatch repair and colon cancer. Adv Exp Med Biol. 2005;570:85–123.
- Taniguchi K, Kakinuma S, Tokairin Y, Ara M, Kohno H, Wakabayashi K, et al. Mild inflammation accelerates colon carcinogenesis in Mlh1-deficient mice. Oncology 2006;71:124–30.
- Bugni JM, Meira LB, Samson LD. Alkylation-induced colon tomorigenesis in mice deficient in the Mgmt and Msh6 proteins. Oncogene 2009;28:734–41.
- Poulogiannis G, Frayling IM, Arends MJ. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010;56:167–79.
- Liao J, Seril DN, Lu GG, Zhang M, Toyokuni S, Yang AL, et al. Increased susceptibility of chronic ulcerative colitis-induced carcinoma development in DNA repair enzyme Ogg1 deficient mice. Mol Carcinog 2008;47:638–46.
- Lu AL, Bai H, Shi G, Chang DY. MutY and MutY homologs (MYH) in genome maintenance. Front Biosci 2006;11:3062–80.
- Casorelli I, Pannellini T, De Luca G, Degan P, Chiera F. , Iavarone I, et al. The Mutyh base excision repair gene influences the inflammatory response in a mouse model of ulcerative colitis. PLoS One 2010;5:e12070.
- Vicario M, Crespi M, Franch A, Amat C, Pelegri C, Moreto M. Induction of colitis in young rats by dextran sulfate sodium. Dig Dis Sci 2005;50:143–50.
- Jung HJ, Kim EH, Mun JY, Park S, Smith ML, Han SS, et al. Base excision DNA repair defect in Gadd45a-deficient cells. Oncogene 2007;26:7517–25.
- Zaky A, Busso C, Izumi T, Chattopadhyay R, Bassiouny A, Mitra S, et al. Regulation of the human AP-endonuclease (APE/Ref-1) expression by the tumor suppressor p53 in response to DNA damage. Nucleic Acids Res 2008;36:1555–66.
- Fung H, Demple B. Distinct roles of Ape1 protein in the repair of DNA damage induced by ionizing radiation or bleomycin. J Biol Chem 2011;286:4968–77.
- Jones LE, Ying L, Hofseth AB, Jelezcova E, Sobol RW, Ambs S, et al. Differential effects of reactive nitrogen species on DNA base excision repair initiated by the alkyladenine DNA glycosylase. Carcinogenesis 2009;30:2123–9.
- Bernstein C, Bernstein H, Payne CM, Garewal H. DNA repair/pro-apoptotic dual role proteins in five major DNA repair pathways: fail-sale protection against carcinogenesis. Mutat Res 2002;511:145–78.
- Ivanovich JL, Read TE, Ciske DJ, Kodner IJ, Whelan AJ. A practical approach to familial and hereditary colorectal cancer. Am J Med 1999;107:68–77.
- Kim MH, Kim HB, Acharya S, Sohn HM, Jun JY, Chang IY, et al. Ape1/Ref-1 induces glial cell-derived neurotropic factor (GDNF) responsiveness by upregulating GDNF receptor α1 expression. Mol Cell Biol 2009;29:2264–77.
- Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal 2009;11;601–20.
- Fujii S, Fujimori T, Kawamata H, Takeda J, Kitajima K, Omotehara F, et al. Development of colonic neoplasia in p53 deficient mice with experimental colitis induced by dextran sulphate sodium. Gut 2004;53:710–6.