Abstract
Objective
Redox imbalance either inside platelets or in their immediate surroundings prove detrimental to their physiologic functions during haemostasis. This study was therefore aimed to assess the effect of peroxide radicals on platelet functions and underlying signalling mechanisms using asparagine-conjugated diperoxovanadate (DPV-Asn).
Methods
Platelet aggregation, ATP secretion, TxB2 release, intra-platelet calcium mobilization, protein tyrosine phosphorylation, GPIIbIIIa activation by PAC1 labelling and sCD40L release (enzyme-linked immunosorbent assay) was monitored using various concentrations of DPV-Asn. Cell viability was assessed by Annexin V labelling, MTT assay, LDH leakage and mitochondrial membrane potential by JC-1.
Results
Platelet aggregation induced by DPV-Asn was chiefly regulated by dense granule secretion, thromboxane A2 (TxA2) generation, intra-platelet [Ca2+] influx, GPIIbIIIa activation and sCD40L release, which were significantly reduced in presence of U73122 (PLC inhibitor), aspirin (COX), SB203580 (p38 inhibitor), and PD98059 (ERK inhibitor). This was further corroborated by enhanced tyrosine phosphorylation of numerous platelet proteins including PLC-γ2, which apparently played a central role in transducing peroxide signals to regulate [Ca2+] influx and phosphorylation of p38 and ERK1/2 MAP kinase.
Discussion
Peroxide radicals critically regulate the thrombo-inflammatory functions of platelets via the PLCγ2-p38-ERK1/2-TxA2 pathway, which closely resembles the clinical scenario of various pathologies like hyperglycemia and atherosclerosis during which oxidative stress disrupts platelet functions.
Introduction
Platelets not only play a central role in haemostasis but also in the pathophysiology of inflammation and atherosclerosis. Reactive oxygen species (ROS) regulate different stages of platelet activation at sites of vascular injury or atherosclerotic plaques as evidenced by both experimental and clinical studies.Citation1–Citation3 Platelets themselves generate ROS during activation Citation4–Citation7 and pharmacologic inhibition by antioxidants regulates platelet activation.Citation8 H2O2, a major cellular oxidant and signalling modulator in mammalian cells,Citation9 induces platelet activation, either at a very high concentration or in the presence of trace element vanadate.Citation10 Moreover, even at a very high concentration of 4 mM, H2O2 failed to trigger any apparent platelet aggregation or tyrosine phosphorylation.Citation10 However, when higher concentration of H2O2 was administered to platelets in combination with vanadate, it formed diperoxovanadate (DPV) complex in solution, which induced a remarkable increase in platelet activation and aggregation.Citation10,Citation11
DPV offers its peroxo groups to participate in peroxidation reactions, and is almost 50 times more stable than H2O2 in the presence of catalase,Citation12,Citation13 thereby requiring several fold lower concentration than H2O2. Previous reports have shown that DPV substitutes H2O2 in a myriad of biological activities such as cell proliferation, insulin mimetic actions, smooth muscle contraction, endothelial functions, as well as modulation of various signalling events such as activation of phospholipase D, PI3-kinase, MAP kinase, intracellular calcium mobilization, tyrosine phosphorylation, and cytoskeletal reorganization.Citation14–Citation18 However, recent finding from our lab has shown that apart from modulating the contraction of rat aortic rings, the peroxovanadate compounds severely compromised the functional integrity of blood vessels.Citation19 The cytotoxic property of DPV was reduced to a minimum when asparagine was coupled to it, thereby forming a novel derivative DPV-Asn. The latter was equally effective to DPV in modulating aortic ring responses, but without any deleterious effects on vascular integrity.
Disturbed flux of ROS either inside platelets or in their immediate surroundings results in alterations in the redox homoestasis, which further proves to be detrimental to their physiologic functions. This study was, thus aimed to investigate the modulation of platelet functions and associated signalling using peroxide mimetic, DPV-Asn. The results reveal the mechanistic insights of the effect of ROS on platelet functions during oxidative stress under various pathological conditions.
Materials and methods
Reagents
Diperoxovanadate-asparagine (DPV-Asn) was prepared by a method described earlier.Citation19 Fibrillar type I collagen from equine tendon and Chrono-Lume® reagent were obtained from Chrono-Log Corp (Pennsylvania, PA, USA). 2′,7′-dichlorofluorescein-diacetate (DCFH-DA), 5,5′-6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazolyl carbocyanine iodide (JC-1), N-acetyl cysteine and Fura-2AM were purchased from Sigma-Aldrich (USA); Hydrogen peroxide (30%) was obtained from Rankem, RFCL Ltd., New Delhi, India (India), Annexin V-FITC kit from BD Biosciences (San Jose, CA, USA); BioTrak thromboxane B2 EIA enzyme immunoassay kit was purchased from Cayman Chemicals (Ann Arbor, MI, USA), human CD40 ligand/TNFSF5 was purchased from R&D Systems (Minneapolis, MI, USA); U73122, PD98059, SB203580 were procured from Calbiochem (La Jolla, CA, USA); anti phospho-Tyrosine antibody 4G10 (Upstate Biotechnology, San Ramon, CA, USA), anti-phospho p38, anti-p38, anti phospho ERK, and anti-ERK1/2 were purchased from Cell Signalling Technology (Danvers, MA, USA), anti-PLC-γ2 antibody was purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Platelet isolation
This study was approved by the Institutional Ethics Committee of CSIR-Central Drug Research Institute, Lucknow and King George's Medical University, Lucknow. Written consent was obtained from all the healthy volunteers and the study was conducted according to the Declaration of Helsinki.
Human blood was collected from healthy volunteers in acid-citrate-dextrose (1:7). Platelet-rich plasma (PRP) was obtained by centrifugation at 180 × g for 10 minutes. For washed platelets, ACD, apyrase type VII (0.5 U/ml) and PGI2 (0.5 µM) were added to PRP and centrifuged at 700 × g for 10 minutes. Platelets were washed in Tyrode's HEPES buffer (134 mM NaCl, 2.9 mM KCl, 1.0 mM MgCl2, 10.0 mM HEPES, 5.0 mM glucose, 12.0 mM NaHCO3, 0.34 mM Na2HPO4; pH 7.4) containing ACD and apyrase and were finally suspended in Tyrode's HEPES buffer (pH 7.4) containing 5 mM glucose.
Measurement of ROS generation
Human platelets (1 × 107 cells/ml) were incubated with vehicle or various inhibitors for 5 minutes at 37°C and subsequently loaded with DCFH-DA (10 µM) for 10 minutes. Finally, DPV-Asn was added at various concentrations to the suspension. Each sample was monitored for free radical generation by acquiring 10 000 cells which were subsequently analysed by Cell Quest program (FACS Calibur; Becton Dickinson, Franklin Lakes, NJ, USA).Citation20
Platelet aggregation and ATP secretion
A turbidimetric method was applied to measure platelet aggregation, using a four channel-Aggregometer (Chrono-Log-Corp, USA).Citation21 PRP, pre-treated with vehicle or pharmacological interventions for 5 minutes, was incubated with DPV-Asn or H2O2 for 10 minutes at 37°C, followed by stirring at 1200 rpm in aggregometer for 5–10 minutes. Simultaneously, ATP secretion was measured using the Chrono-Lume® reagent as per the manufacturer's guidelines. The percentage of aggregation and amount of ATP release was calculated by using Aggrolink Software.
LDH assay
LDH release was measured by spectrophotometric analysis. After incubation with DPV-Asn, the cell suspension was centrifuged and 50 µl aliquots of the resulting supernatant were added to sodium pyruvate (10 mM) and then incubated for 10 minutes at 37°C. Finally 4 mM NADH was added and the decrease in absorbance resulting from conversion of NADH to NAD+ was measured at 340 nm in a UV spectrophotometer. The percentage of total enzyme activity was calculated from control platelets lysed with 0.5% Triton X-100.Citation22
Annexin V labelling
Washed platelets were incubated with DPV-Asn for 15 minutes in presence of 5 µl of Annexin V. Following incubation, 400 µl of binding buffer (10 mM HEPES-NaOH, pH 7.5; 140 mM NaCl; 2.5 mM CaCl2) was added and each sample was subsequently analysed by flow cytometry (FACScalibur, Becton-Dickinson).Citation23
MTT assay
A suspension of washed platelets (3 × 108/ml) was added to a 96-well plate followed by addition of varying concentrations of DPV-Asn. 12 mM stock of MTT (3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added and the samples were incubated at 37°C for 4 hours. The reaction was stopped by adding 50 µl of DMSO, followed by quantification at 540 nm using a microplate spectrophotometer (PowerWave XS2, Biotek, USA).Citation24
Evaluation of mitochondrial membrane potential (Δψm) using JC-1
Washed platelets (1 × 106 cells) were incubated with DPV-Asn for 15 minutes followed by staining with JC-1 (2.5 µg/ml) for 10 minutes.Citation25 The fluorescence signal from JC-1 aggregates (FL-2) or JC-1 monomers (FL-1) was measured and analyzed by flow cytometry (FACScalibur, Becton Dickinson). A decrease in FL-2 fluorescence was represented as loss in mitochondrial transmembrane potential (Δψm).
Thromboxane B2 measurement
Washed platelets were pretreated with vehicle or inhibitors, followed by stimulation with DPV-Asn for 10 minutes at 37°C. The reaction was terminated by adding ice-cold EDTA solution (10 mM) and indomethacin (10 µM), and the samples were centrifuged at 3000 × g for 5 minutes at 4°C. TxB2 was measured in the resulting supernatant using a TxB2 enzyme immunoassay kit.
Measurement of intracellular Ca2+ concentration
PRP was incubated with 5 µM Fura-2 AM for 60 minutes at 37°C. After two washes, platelets were incubated with vehicle or inhibitors for 5 minutes, followed by DPV-Asn for 10 minutes in presence of CaCl2 (2 mM). The changes in Fura-2 fluorescence were measured at dual excitation wavelengths of 340/380 nm and emission wavelength of 510 nm using a fluorescence spectrophotometer (VARIAN, Cary Eclipse). The fluorescence of fully saturated Fura-2 (Fmax) was obtained by lysing the cells with 0.5% triton X-100, while Fmin was determined by adding 10 mM EGTA to the lysed platelets. The changes in intracellular Ca2+ were calculated using the general equation described by Grynkiewicz et al.Citation26
Immunoprecipitation
Washed platelets, pre-incubated with vehicle or inhibitors for 5 minutes, were stimulated with DPV-Asn for 10 minutes. The reaction was arrested by addition of protein extraction buffer (20 mM Tris, pH 7.5, containing 1% TritonX-100, 150 mM NaCl, 1 mM Na3VO4, 1 mM NaF, 1 mM EGTA, 1 mM EDTA, and protease inhibitors) at 4°C for 20 minutes. After centrifugation at 14 000 × g for 15 minutes at 4°C, the supernatants were collected, and the protein concentration was measured using the BCA protein assay reagent kit. A total of 300 µg of platelet protein was incubated overnight with 2 µg of rabbit anti-PLC-γ2 IgG at 4°C after preclearing with equilibrated protein G Sepharose for 1 hour. This was followed by immunoprecipitation with protein G-sepharose conjugate at 4°C for 3 hours. The immune complexes were washed three times with 500 µl of lysis buffer, resuspended in 2 × Laemmli sample buffer, and boiled at 95°C for 5 minutes. The immune complexes were separated by 8% SDS–PAGE and analysed by western blotting.Citation27
Immunoblotting
Washed platelets, pre-incubated with vehicle or inhibitors, were stimulated with DPV-Asn for 10 minutes. The reaction was stopped by the addition of 4X Laemmli Sample loading bufferCitation28 (containing 10 µg/ml aprotinin, 1 mM PMSF, 10 µg/ml leupeptin, 10 mM NaF, 1 mM sodium orthovanadate), followed by boiling for 3 minutes. Samples containing equal amounts of protein (30 µg) were run on 8–10% SDS–PAGE and transferred to nitrocellulose membrane (Amersham Biosciences, Sweden). Blots were probed with antibodies specific for phosphotyrosine, phospho-p38, p38, phospho-ERK and ERK1/2, PLC-γ2 or actin in TBST (Tris 25 mM; pH 7.5, NaCl 150 mM, tween 20 (0.1%)) containing 3% BSA. The membranes were then washed five times for 30 minutes in TBS-T before being incubated with appropriate secondary horseradish peroxidase-conjugated antibody in TBS-T. The proteins were detected using an enhanced chemiluminescence detection system.
sCD40L release
Washed platelets, pre-treated with pharmacologic interventions, were incubated at 37°C with various concentrations of DPV-Asn or collagen for 10 minutes. The samples were centrifuged at 8000 × g for 3 minutes after addition of 10 mM of EGTA and the supernatants were transferred to a clean tube and centrifuged again.Citation29 The supernatants were then immediately tested for sCD40L by enzyme-linked immunosorbent assay as per the manufacturer's instructions.
Statistical analysis
All results are presented as mean ± SEM. One-way analysis of variance (Newman–Keuls’ Multiple or Dunnett's Comparison Test) was used for statistical analysis. A P value of <0.05 was considered to be statistically significant.
Results
DPV-Asn-induced intracellular ROS generation and platelet aggregation
DPV-Asn generated oxidizing radicals inside platelets in a concentration dependent manner. This response remained unhindered in the presence of DPI (NADPH oxidase inhibitor) but was completely abrogated in the presence of N-acetyl cysteine (A). DPV-Asn was found to trigger spontaneous platelet activation and aggregation in a concentration-dependent manner. At 1 µM, DPV-Asn did not induce any platelet aggregation, while at 10 and 25 µM, the mean platelet aggregation was 13 ± 5% and 34 ± 6%, respectively, which was further increased to 68 ± 2% and 75 ± 4% at 50 and 100 µM, respectively. However, H2O2 could induce only 30 ± 4% aggregation even at a very high concentration of 4 mM, while lower concentrations did not induce any measurable platelet aggregation (B).
Figure 1. Effect of DPV-Asn on ROS generation, spontaneous aggregation, cytotoxicity and apoptosis in human platelets. (A) Concentration-dependent (1–100 µM) effect of DPV-Asn on ROS generation assessed by DCFH-DA labelling in human platelets. Washed platelets pre-loaded with DCFH-DA and pre-treated with NAC (5 mM) or DPI (10 µM) for 5 minutes were stimulated with DPV-Asn (50 µM) for 15 minutes. (B) Aggregation profile of platelet-rich plasma stimulated with DPV-Asn (10–300 µM), H2O2 (4 mM, 8 mM) or collagen (1 µg/ml). In (A) and (B), data have been presented as the mean ± SEM of fold change in fluorescence intensity or percent aggregation response of five individual experiments. (C) LDH release was measured in control and DPV-Asn-treated platelets at various concentrations after 1 hour of incubation. (D) MTT toxicity assay was performed on washed platelets to ascertain DPV-Asn toxicity up to 100 µM. In (C) and (D) bar diagrams represent mean ± SEM of three independent experiments. (E) Platelets were incubated with various concentrations of DPV-Asn were labelled with Annexin V-FITC. (F) Mitochondrial membrane potential loss was monitored using the fluorescent probe JC-1. In (E) and (F), dot plots being represented from an individual experiment, which was performed at least three times. *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from control, #P < 0.05 significantly different from DPV-Asn (50 µM)-treated platelets.
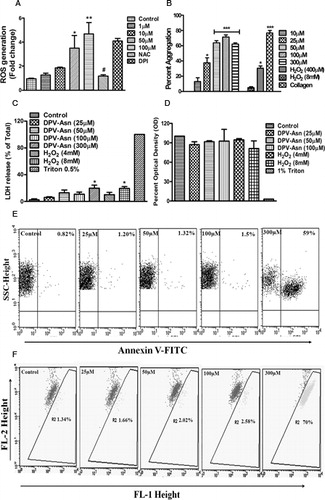
As evidenced from LDH leakage and MTT assay, incubation of platelets with DPV-Asn even at concentration upto 100 µM did not affect the cell viability at either cytosolic or mitochondrial level, respectively (C and D). Moreover, at submaximal and optimal concentrations of DPV-Asn (10–100 µM), sufficient to cause platelet aggregation, there was no indication of PS externalization by Annexin V labelling. However, at a very high concentration of 300 µM, approximately 50% of platelet population became positive for Annexin V and PS exposure (E).
Similarly platelets treated with DPV-Asn (10–100 µM) did not cause any loss in mitochondrial membrane depolarization as evidenced by JC-1 staining, a mitochondrial membrane potential (Ψm) indicator. However, at 300 µM, DPV-Asn caused significant Ψm depolarization (F).
DPV-Asn-induced dense granule secretion in platelets
DPV-Asn stimulated platelets to release ATP in a concentration-dependent manner. At 25 µM, platelets released 0.3 nmoles of ATP which was further augmented to 1.2 and 2.2 nmoles following stimulation with DPV-Asn at 50 and 100 µM, respectively. H2O2 even at a concentration of 4 mM failed to elicit any detectable ATP release under similar conditions. The increase in ATP release was significantly attenuated by N-acetylcysteine (NAC). Similar to aggregation, it was also found that calcium chelator BAPTA (10 µM) and the PLC inhibitor U73122 (1 µM) significantly reduced the secretion responses induced by DPV-Asn (A).
Figure 2. Effect of DPV-Asn on dense granule secretion, thromboxane A2 release, [Ca2+] influx and GPIIbIIIa activation. (A) ATP release from human platelets was assessed using luciferin–luciferase reaction after 10 minutes of incubation with DPV-Asn at various concentrations or platelets pre-treated with N-acetyl cysteine, NAC (5 mM), PLC inhibitor U73122 (1 µM) or BAPTA (10 µM) and stimulated with DPV-Asn (50 µM). (B) DPV-Asn induced release of TxB2 from platelets in a concentration-dependent manner. Levels of TxB2 were assayed in the supernatant of washed platelet suspension after 10 minutes of platelet stimulation. (C) DPV-Asn-induced calcium mobilization in human platelets. Platelet-rich plasma was loaded with Fura-2AM (5 µM) for 60 minutes at RT. Platelets were then washed and measurement of intracellular [Ca2+] was performed in 2 ml aliquots of platelet suspension under continuous stirring. Results being expressed as mean increase in intracellular [Ca2+] concentration (nM ± SEM). (D) Increase in platelet [Ca2+] levels upon DPV-Asn stimulation (50 µM) after pre-treatment with NAC (5 mM), U73122 (1 µM), EGTA (1 mM), or BAPTA (10 µM), followed by addition of 2 mM CaCl2 to detect the [Ca2+]. (E) DPV-Asn-induced platelet activation as assessed by the activation of GPIIbIIIa, which was monitored by PAC1-FITC labelling using flow cytometry. The representative overlay histograms have been presented as the number of events versus PAC-1 binding [(a) Control, (b) DPV-Asn (50 µM), (c) DPV-Asn (100 µM), (d) NAC + DPV-Asn (50 µM), (e) BAPTA + DPV-Asn (50 µM), (f) H2O2 (8 mM)] (F) Bar diagram represents the mean fold change in fluorescence intensity (MFI) relative to unstimulated platelets as shown in (E) from three separate experiments ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from unstimulated control, #P < 0.05 significantly different from DPV-Asn (50 µM)-treated platelets.
![Figure 2. Effect of DPV-Asn on dense granule secretion, thromboxane A2 release, [Ca2+] influx and GPIIbIIIa activation. (A) ATP release from human platelets was assessed using luciferin–luciferase reaction after 10 minutes of incubation with DPV-Asn at various concentrations or platelets pre-treated with N-acetyl cysteine, NAC (5 mM), PLC inhibitor U73122 (1 µM) or BAPTA (10 µM) and stimulated with DPV-Asn (50 µM). (B) DPV-Asn induced release of TxB2 from platelets in a concentration-dependent manner. Levels of TxB2 were assayed in the supernatant of washed platelet suspension after 10 minutes of platelet stimulation. (C) DPV-Asn-induced calcium mobilization in human platelets. Platelet-rich plasma was loaded with Fura-2AM (5 µM) for 60 minutes at RT. Platelets were then washed and measurement of intracellular [Ca2+] was performed in 2 ml aliquots of platelet suspension under continuous stirring. Results being expressed as mean increase in intracellular [Ca2+] concentration (nM ± SEM). (D) Increase in platelet [Ca2+] levels upon DPV-Asn stimulation (50 µM) after pre-treatment with NAC (5 mM), U73122 (1 µM), EGTA (1 mM), or BAPTA (10 µM), followed by addition of 2 mM CaCl2 to detect the [Ca2+]. (E) DPV-Asn-induced platelet activation as assessed by the activation of GPIIbIIIa, which was monitored by PAC1-FITC labelling using flow cytometry. The representative overlay histograms have been presented as the number of events versus PAC-1 binding [(a) Control, (b) DPV-Asn (50 µM), (c) DPV-Asn (100 µM), (d) NAC + DPV-Asn (50 µM), (e) BAPTA + DPV-Asn (50 µM), (f) H2O2 (8 mM)] (F) Bar diagram represents the mean fold change in fluorescence intensity (MFI) relative to unstimulated platelets as shown in (E) from three separate experiments ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from unstimulated control, #P < 0.05 significantly different from DPV-Asn (50 µM)-treated platelets.](/cms/asset/3c9e8454-d559-462c-9409-d704efe90417/yrer_a_11745390_f0002_b.jpg)
DPV-Asn-induced TxB2 generation
Platelets stimulated by DPV-Asn showed increased production of TxB2 (stable metabolite of TxA2) in a concentration-dependent manner. The TxB2 level in control was 100 ± 35 pg/ml which was significantly increased to 400 ± 53 pg/ml after incubating platelets with 50 µM DPV-Asn. TxB2 levels were further increased to 476 ± 37 pg/ml at 100 µM DPV-Asn (B).
DPV-Asn-induced intracellular [Ca2+] mobilization in platelets
In Fura-2 loaded platelets, DPV-Asn induced significant increase in [Ca2+] from 80 to 450 nM. The DPV-Asn induced increase in [Ca2+] was concentration-dependent with maximum increase occurring at 100 µM but it failed to cause any alteration below 25 µM (C). To further assess the role of calcium, the effect of chelating intracellular and extracellular calcium on DPV-Asn induced [Ca2+] was investigated. DPV-Asn induced increase in [Ca2+] was significantly reduced by antioxidant NAC, PLC inhibitor U73122, calcium chelator EGTA, and BAPTA (D).
DPV-Asn-induced platelet GPIIbIIIa activation
DPV-Asn stimulation induced PAC-1 binding in a concentration dependent manner, which was almost completely inhibited in presence of NAC and BAPTA. At 25 µM, DPV-Asn induced 2 ± 0.6-fold increase in PAC-1 labelling, which was further increased to 4.5 ± 0.5 and 6 ± 1-fold at 50 and 100 µM, respectively. However, H2O2 treatment did not induce PAC-1 labelling of platelets unlike that of DPV-Asn (E and F).
DPV-Asn-induced tyrosine phosphorylation in platelets
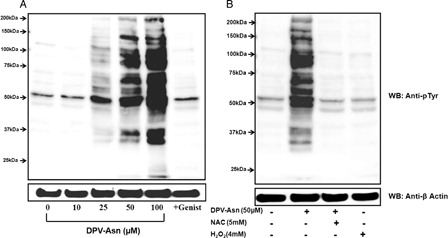
At lower concentration of DPV-Asn (25 µM) only three prominent tyrosine phosphorylated bands between Mr 100–75 kDa, Mr 75–50 kDa and Mr 50 kDa were immunodetected. However, at higher concentrations of DPV-Asn (50–100 µM), additional proteins were detected in the range of Mr ∼150 kDa, and 50–37 kDa. No significant increase in tyrosine phosphorylation of proteins was detected at 10 µM DPV-Asn (A). Moreover, pretreatment of platelets with tyrosine kinase inhibitors genistein (100 µM) attenuated DPV-Asn induced increase in tyrosine phosphorylation of proteins. The DPV-Asn induced increase in protein tyrosine phosphorylation was almost completely attenuated in presence of N-acetyl cysteine thereby suggesting that protein tyrosine phosphorylation was sensitive to DPV-Asn-induced redox imbalance in platelets (B).
Figure 3. DPV-Asn-induced protein tyrosine phosphorylation in human platelets. (A) Washed platelets pre-incubated at 37°C with saline or 100 µM genistein for 5 minutes, were further incubated with different concentrations of DPV-Asn for 10 minutes. (B) Washed platelet incubated with DPV-Asn (50 µM) in the absence or presence of NAC (5 mM) or H2O2 (4 mM). Blot is representative of three independent experiments.
DPV-Asn-induced platelet aggregation and TxB2 release involves the role of PLC, p38, ERK, and COX
In this study, DPV-Asn (50 µM)-induced platelet aggregation (68 ± 2%) was reduced to 4 ± 0.1% in the presence of N-acetyl cysteine, thereby indicating the involvement of oxidizing radicals in inducing platelet activation. Moreover, treatment of platelets with PLC inhibitor U73122 (1 µM), p38 inhibitor SB203580 (10 µM) and ERK inhibitor PD98059 (10 µM) significantly reduced DPV-Asn-induced platelet aggregation to 6 ± 0.5, 39 ± 11 and 19 ± 2%, respectively. Aspirin pre-treatment also inhibited the DPV-Asn-mediated platelet aggregation to 27 ± 3% suggesting the involvement of COX and associated generation of TxB2 (A).
Figure 4. Effect of pharmacological interventions on DPV-Asn-induced platelet aggregation, thromboxane A2 release, PLC-γ2, and MAP kinase phosphorylation. (A) Platelet-rich plasma was incubated with inhibitors (NAC [5 mM], U73122 [1 µM], BAPTA [10 µM], SB203580 [10 µM], PD98059 [10 µM], aspirin [100 µM], for 5 minutes followed by stimulation with DPV-Asn (50 µM) at 37°C in an aggregometer under continuous stirring. Data being expressed as mean ± SEM of percent aggregation response from five independent experiments performed in duplicate for each data point. (B) DPV-Asn induced TxB2 release from human platelets. Levels of TXB2 were assayed in the supernatant of washed platelet suspension in presence or absence of inhibitors. (C, D) DPV-Asn-induced PLC-γ2 phosphorylation. PLC-γ2 was immunoprecipitated with anti-PLC-γ2 and immunoblotted with anti-phosphotyrosine after washed platelets were stimulated with DPV-Asn at various concentrations (25, 50, 100 µM) (C) or washed platelets pre-treated with NAC (5 mM) or U73122 (1 µM) followed by stimulation with DPV-Asn (50 µM) for 10 minutes (D). (E, F) DPV-Asn-induced phosphorylation of p38 and ERK1/2 MAP kinases. Washed platelets were incubated for 10 minutes with indicated inhibitors followed by stimulation with DPV-Asn at 37°C, and immunoblotted with phospho p38, p38, phospho ERK1/2, or ERK1/2 antibodies. All immunoblots in (C), (D), (E), and (F) are representative of three independent experiments under similar conditions. Upper panels represent the mean densitometric scans ± SEM of three experiments. *P < 0.05 significantly different from control, #P < 0.05, ##P < 0.01 significantly different from DPV-Asn (50 µM).
![Figure 4. Effect of pharmacological interventions on DPV-Asn-induced platelet aggregation, thromboxane A2 release, PLC-γ2, and MAP kinase phosphorylation. (A) Platelet-rich plasma was incubated with inhibitors (NAC [5 mM], U73122 [1 µM], BAPTA [10 µM], SB203580 [10 µM], PD98059 [10 µM], aspirin [100 µM], for 5 minutes followed by stimulation with DPV-Asn (50 µM) at 37°C in an aggregometer under continuous stirring. Data being expressed as mean ± SEM of percent aggregation response from five independent experiments performed in duplicate for each data point. (B) DPV-Asn induced TxB2 release from human platelets. Levels of TXB2 were assayed in the supernatant of washed platelet suspension in presence or absence of inhibitors. (C, D) DPV-Asn-induced PLC-γ2 phosphorylation. PLC-γ2 was immunoprecipitated with anti-PLC-γ2 and immunoblotted with anti-phosphotyrosine after washed platelets were stimulated with DPV-Asn at various concentrations (25, 50, 100 µM) (C) or washed platelets pre-treated with NAC (5 mM) or U73122 (1 µM) followed by stimulation with DPV-Asn (50 µM) for 10 minutes (D). (E, F) DPV-Asn-induced phosphorylation of p38 and ERK1/2 MAP kinases. Washed platelets were incubated for 10 minutes with indicated inhibitors followed by stimulation with DPV-Asn at 37°C, and immunoblotted with phospho p38, p38, phospho ERK1/2, or ERK1/2 antibodies. All immunoblots in (C), (D), (E), and (F) are representative of three independent experiments under similar conditions. Upper panels represent the mean densitometric scans ± SEM of three experiments. *P < 0.05 significantly different from control, #P < 0.05, ##P < 0.01 significantly different from DPV-Asn (50 µM).](/cms/asset/29427ce2-9976-4a19-8437-27c4feb2ad9a/yrer_a_11745390_f0004_b.jpg)
This was further confirmed as pre-incubation of platelets with Aspirin (100 µM), U73122 (1 µM) and BAPTA (10 µM) significantly decreased the DPV-Asn-induced TxB2 production. Furthermore, treatment with PD98059 (ERK inhibitor) and SB203580 (p38 inhibitor) also attenuated DPV-Asn-induced TxB2 release suggesting that PLC-γ2 and its downstream effector p38 and ERK MAP kinase contributes to DPV-Asn-induced TxB2 release (B).
DPV-Asn-activated PLC-γ2, ERK, and p38 MAPK in platelets
DPV-Asn induced phosphorylation of PLC-γ2 at tyrosine residue in a concentration-dependent manner as evident in the immunoprecipitates of both control and stimulated platelet lysates (C). Treatment of platelets with PLC inhibitor U73122 or ROS scavenger NAC significantly reduced DPV-Asn-induced phosphorylation of PLC-γ2 (D). Moreover, DPV-Asn also augmented the phosphorylation of both p38 and ERK1/2 (E and F) and pre-treatment of platelets with p38-specific inhibitor SB203580 or ERK1/2-specific inhibitor PD98059, PLC inhibitor U73122, and ROS scavenger NAC significantly inhibited DPV-Asn-induced phosphorylation of p38 and ERK1/2. This further indicated that p38 and ERK1/2 MAPK activation was regulated by PLC-γ2 during DPV-Asn-induced platelet aggregation, which in turn is associated with oxidative stress.
DPV-Asn-induced sCD40L release
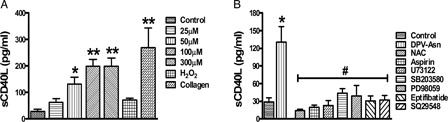
DPV-Asn at 50 µM concentration augmented the release of sCD40L after stimulating platelets for 30 minutes without stirring (130.8 ± 25 pg/ml, compared to control; 29 ± 7.3 pg/ml) (A). This peroxide-dependent release of sCD40L from intra-platelet stores of platelets was significantly inhibited by PLC inhibitor U73122 (67 ± 6%), Aspirin, a COX-1 inhibitor (85 ± 2.5%), or SQ29548 a TP antagonist (76 ± 6%) (B). To further assess whether the activation of MAP kinase p38 and ERK1/2 also contributes to the release of sCD40L, in concert with DPV-Asn-induced platelet aggregation and thromboxane A2 release, the effect of each MAPK specific inhibitor was tested. Compared to control, SB203580, a specific inhibitor of p38 MAPK and PD98059, a specific inhibitor of ERK1/2 significantly suppressed the DPV-Asn-induced sCD40L release by (67 ± 7% and 70 ± 10%), respectively. Moreover, sCD40L release from DPV-Asn stimulated platelets was significantly attenuated in presence of GPIIbIIIa inhibitor Eptifibatide (76 ± 6%), thereby indicating that the release of sCD40L is closely associated with the activation of GPIIbIIIa (B).
Figure 5. DPV-Asn-induced release of sCD40L is regulated by PLC-γ2 and thromboxane A2. (A) DPV-Asn, H2O2 (8 mM) or collagen (6 µg/ml)-induced release of soluble CD40L (sCD40L) from human washed platelets. The soluble CD40L (sCD40L) released from activated platelets in the presence of DPV-Asn, H2O2 or collagen was measured by enzyme-linked immunosorbent assay. (B) Effect of various pharmacologic interventions on DPV-Asn-induced sCD40L release from platelets (NAC (5 mM), aspirin (100 µM), U73122 (1 µM), SB203580 (10 µM), PD98059 (10 µM), eptifibatide (10 µM), SQ29548 (100 nM)). Data have been presented as mean ± SEM of amount of sCD40L released (pg/ml) of four independent experiments (*P < 0.05, **P < 0.01 vs. control platelets).
Discussion
This study was undertaken to revisit the effect of peroxide radicals on platelet activation and signalling so as to correlate with altered responses observed under the conditions of oxidative stress in association with diverse pathologies. We herein show for the first time that peroxide mimetic DPV-Asn activates platelets through PLC-γ2 mediated p38 and ERK1/2 MAP kinase which further drives the release of TxA2 and sCD40L, thereby simulating the functional and molecular mechanisms that operate in platelets during oxidative stress.
Once considered a toxic oxidant, H2O2 has now been accepted to exert regulatory role in diverse cellular functions and acts as an intracellular signalling molecule in mammalian cells. Although H2O2/peroxide radicals play a critical role in modulating platelet reactivity under both physiologic and disease conditions,Citation2,Citation7,Citation30,Citation31 but it very often fails to elicit any measurable platelet aggregation or activation, owing to its susceptibility to degradation by catalase. However, when it reacts with a trace element vanadate, H2O2 forms DPV and induces peroxide-dependent platelet activation as well as protein tyrosine phosphorylation.Citation10,Citation11 Previous report from this lab has shown that DPV, when conjugated with amino acid asparagine (DPV-Asn), preserved the vascular integrity and function of rat aortic rings and hence was found to be safer than DPV alone.Citation19 In this study, the concentrations of DPV-Asn required for platelet activation and signalling, did not induce LDH leakage, PS externalization or loss of mitochondrial membrane potential/integrity in human platelets. However, the unconjugated DPV significantly altered the platelet viability even at relatively lower concentrations. The concentration of DPV-Asn employed to initiate platelet aggregation and secretion was several fold lower than H2O2 alone or the combination of H2O2 and vanadate used in the previous studies.Citation10,Citation11 The cellular antioxidants like catalase and glutathione peroxidase as well as plasma proteins rapidly decompose H2O2 and consequently reduce its potency to aggregate platelets. On the contrary, DPV-Asn enters the cells through ion channels thereby escaping membrane antioxidantsCitation14 which explains its ability to induce a similar degree of platelet activation at a much lower concentration. Even in the absence of stirring, DPV-Asn-induced strong activation of platelet GPIIbIIIa as evidenced from PAC-1 binding, while H2O2 or vanadate alone reportedly induced no such effects.Citation11
While exploring the mechanisms leading to DPV-Asn induced platelet hyperactivity and aggregation, it was observed that PLC-γ2 played a central role in transducing DPV-Asn signals to enhance platelet reactivity involving intra-platelet [Ca2+] mobilization, TxA2 generation and sCD40L release. This is in close agreement with previous clinical studies regarding hyperglycemia-induced oxidative stress in platelets in which the PLC-γ2 phosphorylation was elevated followed by cPLA2-mediated arachidonic acid mobilization which ultimately promoted thromboxane A2 formation.Citation2,Citation27,Citation31 In patients with type II diabetes mellitus and coronary artery disease, oxidative stress governed the release of sCD40L which was directly correlated with enhanced TxA2 production and subsequent activation of MAP kinase and GPIIbIIIa.Citation32,Citation33 The results from this study, therefore indicates that PLC-γ2 unites the pathways involved in the generation of thromboxane A2 and release of sCD40L during DPV-Asn-induced platelet activation.
Many PLC isoforms have been identified in platelets, and an earlier report by Blake et al.Citation10 has shown that platelets treated with unconjugated DPV, formed by the combination of H2O2 (4 mM) and vanadate (400 µM), exhibited tyrosine phosphorylation of a number of proteins including PLC-γ1. However, clinical and experimental evidence suggests that signalling through oxidizing radicals during hyperglycemia and hyperhomocysteinemia is mediated by the selective activation of PLC-γ2.Citation27,Citation31 PLC-γ2 also participates in platelet activation induced by collagen, which itself is correlated with the intracellular generation of H2O2 and superoxide in platelets.Citation6,Citation34 Activation of PLC-γ enzymes involves phosphorylation on tyrosine residues. PLC-γ isoenzymes are highly expressed in platelets and the amount of PLC-γ2 is ∼6-fold higher than that of PLC-β2.Citation35 Earlier studies have shown that collagen-induced PLC activation was dependent on H2O2-mediated tyrosine kinase activation.Citation6 In this study with DPV-Asn, PLC-γ2 phosphorylation was greatly increased in platelets and PLC inhibitor U73122 effectively prevented DPV-Asn-induced ATP release, TxA2 generation and aggregation, suggesting that DPV-Asn-induced tyrosine kinase mediated phosphorylation of PLC-γ2 played a central role in regulating platelet function and signalling. However, the contribution of other PLC isoforms cannot be ignored during DPV-Asn-induced platelet activation and hence their precise contribution during DPV-Asn/peroxide-induced platelet activation should be further investigated. Moreover, DPV-Asn also caused a PLC-dependent rise in [Ca2+] in a concentration-dependent manner which was attenuated in the presence of EGTA, BAPTA, and U73122. Taken together, both intracellular stores and extracellular calcium entry seem to contribute to DPV-Asn-induced calcium increase in platelets. On the contrary, H2O2 has been previously shown to increase [Ca2+]i in human platelets but its half maximal effects were observed at 1 mM concentration.Citation36,Citation37
In this study with DPV-Asn, phosphorylation of PLC-γ2 was inhibited by the antioxidant NAC, thereby suggesting its role in platelet activation during oxidative stress. Apart from PLC-γ2, DPV-Asn also induced phosphorylation of a multitude of proteins at tyrosine residue, which was observed at a remarkably much lower concentration, while H2O2 failed to exhibit any increase even at 4 mM. Tyrosine kinase inhibitor genistein significantly attenuated the enhanced phosphorylation of all the proteins, indicating that the DPV-Asn besides inhibiting protein tyrosine phosphatase, also stimulated tyrosine kinases. Previous studiesCitation10 in which DPV activated PLC and PI-3K by augmenting tyrosine phosphorylation also support the present observations. DPV-Asn also induced the phosphorylation of p38 MAP kinase and ERK1/2, which was significantly attenuated in presence of NAC, calcium chelator BAPTA and PLC inhibitor U73122, suggesting role of these signalling proteins downstream to PLC-γ2. This was in agreement with the earlier reports stating that platelet activation under oxidative stress was regulated by the complex web of these signalling proteins.Citation31,Citation38 Moreover, inhibition of PLC, p38 and ERK significantly attenuated DPV-Asn-induced thromboxane A2 generation and platelet aggregation, thus confirming their role in modulating platelet functions during oxidative stress (summarized in ).
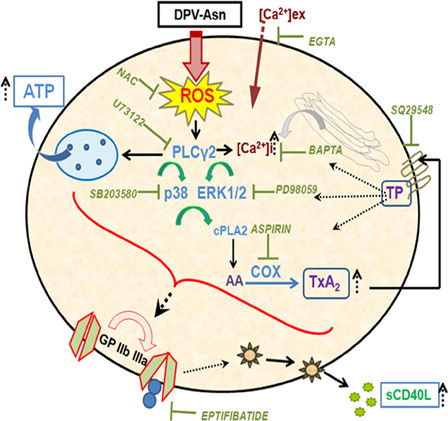
Figure 6. Schematic illustration of DPV-Asn-induced oxidative stress in platelets causing aggregation and sCD40L release. The model depicts that catalase-tolerant peroxide DPV-Asn mediates sequential induction of a signaling cascade in platelets, chiefly regulated by PLC-γ2, which apparently played a central role in upregulating dense granule secretion, calcium influx, p38 and ERK1/2 MAP kinase phosphorylation, and subsequently directed COX activation and enhanced TxA2 generation to further amplify platelet aggregation and thrombus formation. DPV-Asn further augments GPIIbIIIa-dependent release of proinflammatory cytokine sCD40L, thereby exhibiting a thrombo-inflammatory phenotype. DPV-Asn, diperoxovanadate-asparagine; ROS, reactive oxygen species; NAC, N-acetyl cysteine; AA, arachidonic acid; COX, cycloxygenase; TxA2, thromboxane A2; TP, thromboxane-prostanoid receptor.
Apart from the signalling events associated with peroxide-dependent platelet activation, this study also provides the first evidence regarding DPV-Asn-mediated release of sCD40L from platelets in a concentration-dependent manner, which was significantly attenuated in presence of antioxidant NAC. Numerous studies have demonstrated that sCD40L plays a crucial role in cardiovascular complications and is widely used as a prognostic marker for inflammation, thrombosis and atherosclerosis.Citation29,Citation39 It has been shown earlier that platelet activation by collagen and thrombin exhibit enhanced CD40L expression and release, in association with NADPH oxidase (gp91-phox) mediated reactive oxygen intermediates formation.Citation40 Using DPV-Asn as an oxidant to stimulate platelets it was observed that the release of sCD40L from platelets was mediated, at least in part, by PLC-γ2, TxA2 and associated MAP kinase activation followed by subsequent GP IIb/IIIa activation pathway. The involvement of thromboxane A2-dependent signalling during sCD40L release has previously been implicated in atherosclerotic patients with stenosed vessels.Citation32,Citation41 However, our study highlights the contribution of PLC in regulating sCD40L release during oxidative stress, which should be further substantiated with more elaborate experimental and clinical studies.
This study demonstrates that peroxide radical if available in sufficient amounts, is a strong activator of thrombo-inflammatory complications. It will be worthwhile to investigate its additive or synergistic effects on platelet activation with platelet activators such as ADP, thrombin, and collagen. Moreover, since ROS have been widely implicated in the pathophysiology of a number of vascular disorders including atherosclerosis, thrombosis, and diabetes mellitus, a better understanding of the mechanisms of oxidant-stimulated signal transduction pathways might provide novel insights into functions of various cells under normal and pathological conditions.
Conclusion
It can be therefore summarized that the catalase-tolerant peroxide, DPV-Asn stimulated platelets by activating the key signalling proteins like PLC-γ2, which apparently played a central role in not only upregulating dense granule secretion and calcium influx but also activating downstream p38 and ERK1/2 MAP kinase signalling. The p38 and ERK1/2 MAP kinase further directed COX activation via cPLA2 and enhanced thromboxane A2 generation to further amplify platelet aggregation and thrombus formation. Moreover, apart from disrupting the haemostatic functions of platelets, DPV-Asn also augmented the release of proinflammatory cytokine sCD40L thereby leading to a thrombo-inflammatory phenotype. DPV-Asn apart from being useful as insulin mimetic, might also serve as an effective tool to generate oxidative stress in cellular systems to mimic various pathophysiologic conditions.
Funding
The study was supported by a financial grant to Madhu Dikshit from the CSIR project THUNDER (THU0001) and Department of Biotechnology-INDIGO project (GAP0073), India. The award of research fellowship to Ankita Misra from the Council of Scientific and Industrial Research (CSIR), New Delhi, India is duly acknowledged.
Acknowledgements
The authors are grateful to Dr T. Ramasarma (Department of Biochemistry, Indian Institute of Sciences, Bangalore, India) for his excellent guidance regarding understanding of peroxovanadate chemistry. The excellent technical help of Mr A.L. Vishwakarma and Mrs M. Chaturvedi for the Flow Cytometry study, Sophisticated Analytical Instrument Facility (SAIF); CSIR-Central Drug Research Institute, Lucknow, is duly acknowledged.
References
- Violi F, Pignatelli P. Platelet oxidative stress and thrombosis. Thromb Res 2012;129(3):378–81.
- Iuliano L, Colavita AR, Leo R, Pratico D, Violi F. Oxygen free radicals and platelet activation. Free Radic Biol Med 1997;22(6):999–1006.
- Sill JC, Proper JA, Johnson ME, Uhl CB, Katusic ZS. Reactive oxygen species and human platelet GP IIb/IIIa receptor activation. Platelets 2007;18(8):613–9.
- Leoncini G, Maresca M, Colao C. Oxidative metabolism of human platelets. Biochem Int 1991;25(4):647–55.
- Maresca M, Colao C, Leoncini G. Generation of hydrogen peroxide in resting and activated platelets. Cell Biochem Funct 1992;10(2):79–85.
- Pignatelli P, Pulcinelli FM, Lenti L, Gazzaniga PP, Violi F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood 1998;91(2):484–90.
- Bakdash N, Williams MS. Spatially distinct production of reactive oxygen species regulates platelet activation. Free Radic Biol Med 2008;45(2):158–66.
- Sobotkova A, Masova-Chrastinova L, Suttnar J, Stikarova J, Majek P, Reicheltova Z, et al. Antioxidants change platelet responses to various stimulating events. Free Radic Biol Med 2009;47(12):1707–14.
- Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006;312(5782):1882–3.
- Blake RA, Walker TR, Watson SP. Activation of human platelets by peroxovanadate is associated with tyrosine phosphorylation of phospholipase C gamma and formation of inositol phosphates. Biochem J 1993;290(Pt 2):471–5.
- Irani K, Pham Y, Coleman LD, Roos C, Cooke GE, Miodovnik A, et al. Priming of platelet alphaIIbbeta3 by oxidants is associated with tyrosine phosphorylation of beta3. Arterioscler Thromb Vasc Biol 1998;18(11):1698–706.
- Ravishankar HN, Rao AV, Ramasarma T. Catalase degrades diperoxovanadate and releases oxygen. Arch Biochem Biophys 1995;321(2):477–84.
- Rao AV, Ravishankar HN, Ramasarma T. Diperoxovanadate participates in peroxidation reactions of H2O2 in presence of abundant catalase. Biochim Biophys Acta 1998;1381(2):249–55.
- Chatterjee N, Kiran S, Ram BM, Islam N, Ramasarma T, Ramakrishna G. Diperoxovanadate can substitute for H2O2 at much lower concentration in inducing features of premature cellular senescence in mouse fibroblasts (NIH3T3). Mech Ageing Dev 2011;132(5):230–9.
- Natarajan V, Vepa S, Shamlal R, Al-Hassani M, Ramasarma T, Ravishankar HN, et al. Tyrosine kinases and calcium dependent activation of endothelial cell phospholipase D by diperoxovanadate. Mol Cell Biochem 1998;183(1–2):113–24.
- Usatyuk PV, Fomin VP, Shi S, Garcia JG, Schaphorst K, Natarajan V. Role of Ca2+ in diperoxovanadate-induced cytoskeletal remodeling and endothelial cell barrier function. Am J Physiol Lung Cell Mol Physiol 2003;285(5):L1006–17.
- Natarajan V, Scribner WM, Morris AJ, Roy S, Vepa S, Yang J, et al. Role of p38 MAP kinase in diperoxovanadate-induced phospholipase D activation in endothelial cells. Am J Physiol Lung Cell Mol Physiol 2001;281(2):L435–49.
- Parinandi NL, Roy S, Shi S, Cummings RJ, Morris AJ, Garcia JG, et al. Role of Src kinase in diperoxovanadate-mediated activation of phospholipase D in endothelial cells. Arch Biochem Biophys 2001;396(2):231–43.
- Khanna V, Jain M, Barthwal MK, Kalita D, Boruah JJ, Das SP, et al. Vasomodulatory effect of novel peroxovanadate compounds on rat aorta: role of rho kinase and nitric oxide/cGMP pathway. Pharmacol Res 2011;64(3):274–82.
- Sharma P, Barthwal MK, Dikshit M. NO synthesis and its regulation in the arachidonic-acid-stimulated rat polymorphonuclear leukocytes. Nitric Oxide 2002;7(2):119–26.
- Prakash P, Misra A, Surin WR, Jain M, Bhatta RS, Pal R, et al. Anti-platelet effects of Curcuma oil in experimental models of myocardial ischemia-reperfusion and thrombosis. Thromb Res 2011;127(2):111–8.
- Kim EJ, Lim KM, Kim KY, Bae ON, Noh JY, Chung SM, et al. Doxorubicin-induced platelet cytotoxicity: a new contributory factor for doxorubicin-mediated thrombocytopenia. J Thromb Haemost 2009;7(7):1172–83.
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995;184(1):39–51.
- Steele BM, Harper MT, Macaulay IC, Morrell CN, Perez-Tamayo A, Foy M, et al. Canonical Wnt signaling negatively regulates platelet function. Proc Natl Acad Sci USA 2009;106(47):19836–41.
- Jobe SM, Wilson KM, Leo L, Raimondi A, Molkentin JD, Lentz SR, et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008;111(3):1257–65.
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985;260(6):3440–50.
- Leoncini G, Bruzzese D, Signorello MG. A role for PLCgamma2 in platelet activation by homocysteine. J Cell Biochem 2007;100(5):1255–65.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227(5259):680–5.
- Pignatelli P, Sanguigni V, Lenti L, Loffredo L, Carnevale R, Sorge R, et al. Oxidative stress-mediated platelet CD40 ligand upregulation in patients with hypercholesterolemia: effect of atorvastatin. J Thromb Haemost 2007;5(6):1170–8.
- McNicol A, Robertson C, Gerrard JM. Vanadate activates platelets by enhancing arachidonic acid release. Blood 1993;81(9):2329–38.
- Tang WH, Stitham J, Gleim S, Di Febbo C, Porreca E, Fava C, et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J Clin Invest 2011;121(11):4462–76.
- Enomoto Y, Adachi S, Matsushima-Nishiwaki R, Doi T, Niwa M, Akamatsu S, et al. Thromboxane A(2) promotes soluble CD40 ligand release from human platelets. Atherosclerosis 2010;209(2):415–21.
- Santilli F, Davi G, Consoli A, Cipollone F, Mezzetti A, Falco A, et al. Thromboxane-dependent CD40 ligand release in type 2 diabetes mellitus. J Am Coll Cardiol 2006;47(2):391–7.
- Caccese D, Pratico D, Ghiselli A, Natoli S, Pignatelli P, Sanguigni V, et al. Superoxide anion and hydroxyl radical release by collagen-induced platelet aggregation–role of arachidonic acid metabolism. Thromb Haemost 2000;83(3):485–90.
- Lee SB, Rao AK, Lee KH, Yang X, Bae YS, Rhee SG. Decreased expression of phospholipase C-beta 2 isozyme in human platelets with impaired function. Blood 1996;88(5):1684–91.
- Rosado JA, Redondo PC, Salido GM, Gomez-Arteta E, Sage SO, Pariente JA. Hydrogen peroxide generation induces pp60src activation in human platelets: evidence for the involvement of this pathway in store-mediated calcium entry. J Biol Chem 2004;279(3):1665–75.
- Redondo PC, Salido GM, Rosado JA, Pariente JA. Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms. Biochem Pharmacol 2004;67(3):491–502.
- Coulon L, Calzada C, Moulin P, Vericel E, Lagarde M. Activation of p38 mitogen-activated protein kinase/cytosolic phospholipase A2 cascade in hydroperoxide-stressed platelets. Free Radic Biol Med 2003;35(6):616–25.
- Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998;391(6667):591–4.
- Pignatelli P, Sanguigni V, Lenti L, Ferro D, Finocchi A, Rossi P, et al. gp91phox-dependent expression of platelet CD40 ligand. Circulation 2004;110(10):1326–9.
- Furman MI, Krueger LA, Linden MD, Barnard MR, Frelinger AL, Michelson AD. Release of soluble CD40L from platelets is regulated by glycoprotein IIb/IIIa and actin polymerization. J Am Coll Cardiol 2004;43(12):2319–25.