Abstract
Mammalian cells produce reactive oxygen and nitrogen species (ROS/RNOS) in response to an oxidative environment. Powerful antioxidant mechanisms have been developed in order to avoid oxidative stress by contributing to the maintenance of redox homeostasis. Traditionally, accumulation of ROS/RNOS is considered deleterious for cells as it can lead to loss of cellular function, aging, and cell death. Consequently, ROS/RNOS imbalance has been implicated in the etiology and/or progression of numerous pathologies such as cardiovascular diseases, inflammation, and cancer. An interesting concept that has emerged more recently is that not only have cells developed efficient systems to cope with ROS/RNOS accumulation but they have also learned to profit of them under certain circumstances. This notion is supported by data showing that ROS/RNOS can act as signaling molecules affecting the function and activity of a multiplicity of protein kinases and phosphatases controlling cellular homeostasis. This review does not provide an exhaustive overview of molecular mechanisms linked to ROS/RNOS generation and processing but includes relevant examples highlighting the dichotomic nature of these small molecules and the multitude of effects elicited by their accumulation. This aspect of ROS/RNOS ought to be taken into account particularly in novel therapeutic setups that aim to achieve high efficiency and minimal or no side effects.
Introduction
What are ROS/RNOS?
Reactive oxygen species (ROS) are small molecules or ions containing oxygen that either are/ or can be converted into highly reactive molecules/ions. Some of them are free radicals, i.e. they contain an unpaired electron. Examples of ROS are the hydroxyl radical (OH·), superoxide ion (O2−·), and hydrogen peroxide (H2O2). Reactive oxygen-nitrogen species (RNOS) are compounds similar to ROS, which also contain nitrogen. Examples of RNOS are nitrogen oxide (NO·), nitrogen dioxide radical (NO2·), and peroxynitrite (OONO−).
Reactivity of ROS/RNOS
ROS or RNOS are generally oxidants, i.e. in a physiological context they are capable of oxidizing a number of biological molecules such as DNA, proteins, and lipids resulting in damage to the cell, which again may lead to cell malfunction and death. The most reactive ROS is the hydroxyl radical, which can oxidize even very unreactive molecules such as alcohols:with the formation of an alkoxy radical. This can initiate a chain reaction. Other ROS, e.g. H2O2, and superoxide are not strong oxidants, but they can undergo reactions which lead to the formation of the hydroxyl radical. Two such physiologically relevant reactions are the Haber–Weiss reaction and Fenton's reaction, respectively.Citation1 In the Haber–Weiss reaction hydrogen peroxide accepts an electron from superoxide, which leads to the formation of OH·:
Fenton's reaction is very similar to the Haber–Weiss reaction but here ferrous ion (Fe2+) is the electron donor:
The hydroxyl radical can readily react with most biological compounds and lead to damage of lipids, proteins and nucleic acids.Citation2 On the other hand, its reactivity is so high that damage will be limited to a small neighborhood around its source.Citation3 Most other ROS/RNOS, except for peroxynitrite, are only mild oxidants; some of them, e.g. H2O2, can diffuse relatively long distances before they are degraded.
How are ROS/RNOS formed?
Some ROS/RNOS are formed as by-products of physiological reactions, while others are formed in physiologically regulated reactions. In recent years, it has become increasingly difficult to distinguish these two processes: what was originally seen as an unwanted malicious process turns out to be beneficial to the organism in some situations.Citation4 In any case, it is generally acknowledged that a major source of ROS is the one-electron reduction of oxygen to superoxide by reactions in the mitochondrial electron transport chain, primarily by complex I (ubiquinone: NADH oxidoreductase) and by the semiquinone of ubiquinone (coenzyme Q).Citation5 The superoxide formed in this process is rapidly converted into hydrogen peroxide through the action of superoxide dismutase (SOD):
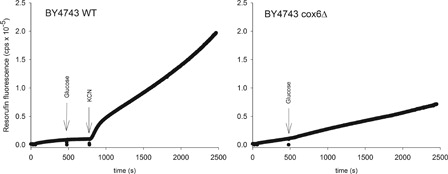
In normal mitochondria formation of ROS is rather slow, but in damaged or aging mitochondria significant amounts of ROS can be produced.Citation5 To illustrate this, we show ROS production in two strains of the yeast Saccharomyces cerevisiae BY4743 (). In the wild-type strain ROS production is low when respiration is turned on by addition of glucose. ROS production increases dramatically after addition of KCN, which blocks respiration. When respiration is blocked electrons accumulate at complex I and on ubiquinone, which then facilitates reduction of oxygen to superoxide. In the cox6Δ strain respiration is blocked from the beginning due to the lack of functional cytochrome c oxidase and ROS production is always high.
Figure 1. ROS production from mitochondria in the wild-type Saccharomyces cerevisiae strain BY4743 (left) and in an isogenic strain where the gene encoding the subunit Cox6p of cytochrome c oxidase has been deleted (right). The latter strain is respiratory incompetent. ROS is determined as hydrogen peroxide using an amplex red peroxidase assay.72 Resorufin fluorescence results from oxidation of amplex red by H2O2, catalyzed by peroxidase. In the BY4743 wild-type strain ROS production is very low following onset of respiration by glucose, but increases dramatically following addition of cyanide, which inhibits respiration 100%. In the cox6Δ mutant strain ROS production is always high. Both strains were starved for three hours before the experiment in order to empty the cells for endogenous substrate.
Some ROS are also produced in reactions catalyzed by xanthine oxidase (XO) and cytochrome P450. Xanthine oxidase exists in two forms, a xanthine dehydrogenase, which uses NAD+ as electron acceptor and a post translational form of this enzyme, which instead of NAD+ uses molecular oxygen as the electron acceptor. The latter leads to formation of superoxide, and it has been proposed that the oxidase form of XO participates in oxidant-mediated signaling in cells.Citation6 Hydrogen peroxide and superoxide are also intermediates in certain peroxidase-catalyzed reactions.Citation7
The formation of RNOS begins with the synthesis of nitrogen oxide, NO·, from arginine, oxygen, and NADPH, catalyzed by NO synthase.Citation8 Nitrogen oxide is itself not very reactive, but is one of the few stable free radicals that exist. However, in the presence of superoxide NO· will be transformed into peroxynitrite, which is a strong oxidizing agent:
Peroxynitrite may cause strand breaks of DNA and it causes irreversible damage to mitochondrial complexes.Citation8 Most of the biological production of peroxynitrite occurs in phagocytic cells in the immune system.
Production of ROS is not always an unwanted and potentially harmful process. In phagocytic immune cells massive amounts of ROS are formed through the concerted action of two enzymes: inducible nitrogen oxide synthase and the membrane-bound NADPH oxidase (NOX). The latter catalyzes the reaction:
The function of ROS produced by phagocytic cells is to kill bacteria and other invading microorganisms.Citation9,Citation10 They are either released into the extracellular space or into the specialized phagolysosome in which the microorganism has been engulfed. The superoxide formed in the NOX-catalyzed reaction may either react with NO to form peroxynitrite or with hydrogen peroxide formed by dismutation of superoxide (catalyzed by SOD) in the Haber–Weiss reaction to produce hydroxyl radical. These ROS species rapidly inactivate the microorganism's proteins, lipids, and DNA. NOX is also present in smaller amounts in normal cells. Here the ROS formed are believed to have a signaling role.Citation11,Citation12
Antioxidant enzymes
A number of enzymes serve to regulate the amount of ROS in the cell. One of these is superoxide dismutase, mentioned above. SOD is predominantly localized in the cytoplasm, in the extracellular space and in the mitochondrial matrix. SOD exists in various forms depending on the central metal ion(s) in the catalytic center, which may either be copper and zinc (Cu-Zn-SOD) or manganese (Mn-SOD). Especially, the latter form (Mn-SOD) has received attention in recent years due to its possible role in cell signaling.Citation13 The hydrogen peroxide formed can be removed in a reaction catalyzed by glutathione peroxidase:Citation14
Here the cysteine-containing tripeptide glutathione (GSH) is oxidized to its disulphide (GS-SG). The disulphide is reconverted into monomeric glutathione by the NADPH-dependent reaction catalyzed by glutathione reductase. Some hydrogen peroxide is also removed in the reaction catalyzed by catalase. However, while catalase is mainly located in the peroxisomes, glutathione peroxidase is found in the cytoplasm and in the mitochondrial matrix. Cells also contain the unusual antioxidant proteins known as peroxiredoxins, which contain active site cysteins. These cysteins undergo oxidation by hydrogen peroxide and are themselves reduced back to sulfhydryl groups by a thioredoxin/thioredoxin reductase system.Citation15 Peroxiredoxins may act as regulators of peroxide concentration in the cells as well as sensors of local peroxides. It has been estimatedCitation3 that the action of peroxiredoxins may reduce the average diffusive length of peroxides in cells from about 1.5 mm to less than 10 µm. This may drastically reduce the damage caused by these ROS. Thus, peroxiredoxins may have significant influence on the amount of ROS/RNOS formed in a tissue.
Role of oxidative stress in life and death
Aerobic organisms make use of detoxification mechanisms in order to eliminate the partially reduced oxygen forms and reactive nitrogen species that are produced during respiration and enzymatic activities. Excessive production and/or accumulation of ROS/RNOS in cells leads to a non-homeostatic state referred to as oxidative- or nitrosative stress as it results in the progressive degradation and/or accumulation of damaged macromolecules such as proteins, lipids, and carbohydrates as mentioned above.Citation16 Although ROS/RNOS at high levels may lead to cell death by damaging organelles, particularly the mitochondria, at low levels they can act as signaling molecules.Citation4 Initial observations supporting the role of ROS/RNOS in signaling regulation derive from studies with human cells showing that for growth factors such as PDGF and EGF, receptor binding was followed by a burst of ROS production and that suppression of ROS rise was accompanied by inhibition of tyrosine kinase signaling induced by ligand binding.Citation17,Citation18 Beside receptor kinases and phosphatases, other signaling molecules appear to be regulated by ROS such as the large family of MAPKs and members of the NF-kB signaling pathway,Citation19 however, the list of potential signaling molecules targeted by ROS is far from being complete.
ROS/RNOS and induction of cell death
Oxidative damage induced by ROS is associated with induction of cell death. Briefly, three major types of cell death can be activated in cells: apoptosis, necrosis, and autophagy.Citation20,Citation21 Apoptosis represents a highly regulated form of cell death that contributes to tissue homeostasis under normal physiological conditions but also to disease pathogenesis. Two major pathways control this process, the extrinsic and intrinsic apoptotic pathways. The former is induced in response to activation of cell surface receptors by death ligands (e.g. Fas and TNF) while the latter is triggered by release of cytochrome c from mitochondria, responding to environmental and chemical stress agents.Citation22–Citation24 Necrosis is a form of cell death that is associated with extensive cell injury causing leakage of cell constituents into the extracellular space that may lead to inflammation and damage of the surrounding tissue. Apoptosis and necrosis are not mutually exclusive events: necrotic death can often develop as a form of late apoptosis that occurs as a result of extensive and prolonged cell damage and partially executed apoptotic program.Citation25 The third major form of cellular death is autophagy, also known as type II programmed cell death, which is essential for cell homeostasis and adaptation to changes in the environment where damaged cell components are degraded and recycled.Citation21 Although autophagy has been recognized as an essential physiological process in healthy cells, the increased formation of autophagic vesicles in dying cells indicates that this process is involved in cell death either as a cause or a consequence of it.
Recently, it has become apparent that elevated levels of hydroxyl radical and superoxide anion play a major role in cell apoptosis through oxidative damage not only of mitochondrial DNACitation26 but also nuclear DNA ().Citation27 Data from Zwacka and co-workers showed that inhibitors of SOD stimulate programmed cell death while overexpression of Mn-SOD and Cu-Zn-SOD reduces apoptosis, suggesting that superoxide anions play an important role in this process.Citation28 Accumulation of ROS appears to be important in apoptosis induction through activation of death receptors. Initial studies carried out employing various model systems showed that antioxidants can block apoptosis induced by TNF and Fas-receptor stimulation, respectively.Citation29,Citation30 More recently, it has been shown that ROS and nitrogen oxide accumulation leads to c-FLIP down-regulation and Fas-induced apoptosis.Citation31 The mode by which ROS stimulate mitochondria-mediated apoptosis is not entirely clear; however, release of cytochrome c seems to be the central event in the initiation of the pro-apoptotic signaling cascade. In this respect, oxidation of the mitochondrial phospholipid cardiolipin has been shown to lead to its decreased binding affinity to cytochrome c, facilitating the mobilization of the latter from the inner mitochondrial membrane (reviewed in ref. Citation32). Other components of the apoptotic machinery such as Bax and Bak contribute to increasing the permeability of the outer mitochondrial membrane by direct interaction with a voltage-dependent anion channel, which positively affects the pore size to an extent sufficient for cytochrome c to be released into the cytoplasm (reviewed in ref Citation32). Like cytochrome c, AIF becomes apoptogenic once released into the cytoplasm and further translocates into the nucleus contributing to induction of a caspase-independent mechanism.Citation33
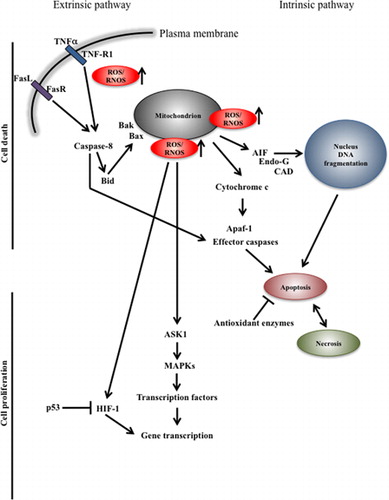
Figure 2. Schematic representation of pro-survival and cell death mechanisms in oxidative stress, respectively. Complex regulatory signals lead to cell death or proliferation. Cell decision is taken on the base of the levels and activity of antioxidant enzymes, level of ROS/RNOS and location. R, receptor; TNF, tumor necrosis factor; ASK1, apoptosis signal-regulating kinase 1; AIF, apoptosis inducing factor; MAPK, mitogen-activated protein kinase; CAD, caspase-activated DNase; Endo-G, endonuclease G; Apaf-1, apoptotic protease activating factor 1; HIF-1, hypoxia-inducible factor 1.
Starvation enhances ROS levels and stimulates autophagy and both superoxide anion and hydrogen peroxide have been shown to mediate induction of autophagy.Citation34–Citation36 One of the few known mechanisms linking redox signaling to autophagy derives from studies with human cancer cell lines showing that enhanced hydrogen peroxide directly regulates the cysteine protease Atg4 by thiol reduction. This process enables the conversion of the cytosolic form of microtubule-associated protein 1A/1B-light chain 3 (LC3-I) into LC3-II (i.e. the form recruited to autophagosomal membranes) and its lipidation, indicative of autophagic activity.Citation34 The effect of nitrogen oxide accumulation on autophagy regulation is not clear to date as it seems to exert both inhibitory and activating effects depending on the cell type.Citation37,Citation38 Finally, increasing evidence suggests that autophagy plays a role in removing oxidatively-damaged molecules as well as in the accumulation of ROS/RNOS. The specific signaling mechanisms controlling whether autophagy should protect or destroy cells awaits further investigation and continues to be an important topic in present and future investigations.
ROS-mediated proliferation signals
Accumulation of ROS is conventionally linked to cytotoxic effects in cells. However, because of the numerous signaling molecules, particularly protein kinases and phosphatases, affected by ROS accumulation it is not surprising that ROS may exert a survival role in cells. This duality is particularly important with respect to some types of diseases such as cancer, where increased ROS production has been linked to resistance of cells to specific anti-neoplastic therapy.Citation39,Citation40 ROS-mediated proliferation signals seem to be partially mediated by members of the MAPK familyCitation41 and also HIF-1α, as the latter has been found to be stabilized by ROS-mediated hydroxylation conferring survival of cancer cells in conditions of limited oxygen availability ().Citation42 An interesting aspect in oxidative studies is that cellular stress does not derive solely from ROS overproduction but also from inactivation or low levels of anti-oxidant enzymes. In this respect, decreased expression levels of mitochondrial Mn-SOD was reported in pancreatic and ovarian cancer and certain subtypes of colorectal carcinomas accompanied by accumulation of superoxide ions.Citation43–Citation45 The opposite is also true. Enhanced levels of Mn-SOD have been reported in primary ovarian cancer and in the blood of patients with leukemia.Citation46,Citation47 As concomitant oxidative stress was observed, this suggests that altered expression of Mn-SOD might be an adaptive response to high oxidant levels or that the antioxidant enzyme is unable to counteract accumulation of ROS. It also underlines the complex regulation of redox balance, which has to be taken into account in the designing of novel therapeutic strategies.
ROS signaling under hypoxia in cancer and ischemia
The concept that many diseases are associated with increased levels of ROS generated by mitochondria is consistent with a ROS-mediated signaling mechanism.Citation48
The transcription factor HIF plays a critical role in the mammalian response to oxygen levels and transcriptionally activates a plethora of genes associated with angiogenesis in cancer, exercise, and ischemia, as well as energy metabolism, nutrient transport, cell cycle and cell migration.Citation49,Citation50 Hence, ΗΙF can be considered a master switch for hypoxic gene expression.Citation49,Citation51 Under normoxia HIF-1α is undetectable. Under hypoxia it rapidly accumulates in the cell nucleus and triggers gene expression.
At normal oxygen concentrations, a set of enzymes, known as prolyl hydroxylases (PHD), that utilize oxygen as a substrate and Fe2+ as a cofactor hydroxylate the proline residues (Pro402 and Pro564) on HIF-1α.Citation52–Citation54 Hydroxylation of HIF-1α at either of the two proline residues creates a recognition site for the binding to the von Hippel-Lindau (pVHL) protein, which has E3 ubiquitin ligase activity and leads to degradation of HIF-1α.Citation55 Under hypoxic conditions, mitochondrial complex III may produce ROS, and the presence of high ROS concentrations has been shown to stabilize HIF-1α,Citation42,Citation56 inasmuch that hydrogen peroxide oxidizes Fe2+ to Fe3+ thus interfering with the necessary binding of Fe2+ to HIF-1α-PHDs.Citation48,Citation57 Additionally, 2-oxoglutarate and succinate are also compounds involved in HIF-1α hydroxylation whose concentrations could be altered by free radicals.Citation58–Citation60 Another mechanism by which ROS could influence the HIF-1 pathway is through changing the availability of oxygen to bind directly to the PHDs or changing PHDs phosphorylation.Citation48
HIF-1α central regulator of oxygen
Cancer – Low oxygen (hypoxia) plays a specific role in solid tumors cells once the size of 1–2 mm has been reached, because at this stage cellular waste products and availability of nutrition become critical for the survival of the tumor cells. At this status HIF-1α production is initiated leading to the transcription of more than 100 new genes, which ensure that the tumor can become angiogenic and thereby survive. Hence, in solid tumors hypoxia is pro-survival (i.e. anti-apoptotic). Yet, there are conflicting reports published on whether hypoxia increases or decreases cellular ROS. Although a number of reports demonstrated increases of ROS under hypoxia from mitochondrialCitation61,Citation62 or non-mitochondrial sources,Citation63 numerous studies have reported that decreased oxygen availability in hypoxia leads to lower ROS concentrations.Citation64,Citation65
A recent reportCitation66 used approaches that specifically changed mitochondrial oxygen consumption rates without affecting ROS production. The results obtained support the notion that HIF-1α protein stabilization in hypoxia occurs independently of mitochondrial ROS production and that mitochondria can modulate the cellular hypoxic response through altered respiratory activity, likely by regulating the cellular oxygen availability. Given that 95% of all cellular oxygen is consumed by mitochondria, it sounds plausible that the mitochondrial metabolic activity in a cell is a major factor in regulating the cellular hypoxic response.
Ischemia – What is observed under hypoxia in tumor cells contrasts to what is seen in normal tissue, e.g. heart tissue under ischemia. As blood flow is reduced to the heart oxygen consumption increases. When the tissue is unable to extract adequate oxygen, the partial pressure of oxygen within the tissue falls (hypoxia) leading to a reduction in mitochondrial respiration and oxidative metabolism. If the hypoxic state is prolonged, cellular death may occur. Here, hypoxia is leading to apoptosis/necrosis (i.e. pro-apoptotic).
In tumor cells, the duration of hypoxia is generally longer than under ischemia, and the cells adapt to anaerobic metabolism and likely to relative stable levels of ROS production. In the normal ischemic cell, the duration of ischemia is often only hours or less and cells are starved of oxygen but unable to adapt as readily as a cancer cell, and furthermore the levels of ROS rapidly increase following the infarct or occlusion and then remain elevated for days.Citation48
The mechanisms for cardiotoxicity may be multifactorial,Citation67 including the impairment of the mitochondrial energetics by increases in the mitochondrial calcium and ROS leading to oxidative stress, cell necrosis, and induction of pro-apoptotic signaling pathways.Citation68
The importance of lack of oxygen in tumors and under ischemia has been traditionally considered separately in basic oncology and cardiology. However, at the bedside the application of pro-apoptotic drugs for the treatment of cancer patients may be contra-indicative with respect to ischemia and therefore this problem has to be seriously taken into consideration in therapeutic treatment.
An example is doxorubicin (DOX), a potent anthracycline antibiotic that has been used in cancer treatments since the late 1960s. However, its clinical use has been hampered by the adverse cardiac effects.Citation69,Citation70 Especially, under long-term treatment DOX can result in the development of cardiomyopathy and congestive failure in a process that involves multiple factors, including the generation of free radicals that damage cellular membranes, disturbance of adrenergic function, alterations in intracellular Ca2+ homeostasis, myocardial cell apoptosis, and selective inhibition of the expression of cardiac muscle-specific proteins.Citation71 Hence, DOX can be considered a janus-faced drug. The increasing knowledge about how ROS signaling pathways work will be helpful to understand the way drugs work for treating for example cancer, but by the same token great care has to be taken to avoid negative influences on healthy organs of patients with diseases other than cancer.
Taking everything into consideration, we would like to sound a note of caution when pro-apoptotic drugs are used to initiate apoptosis in cancer with respect to their potential adverse effect under ischemia.
References
- Mladenka P, Simůnek T, Hübl M, Hrdina R. The role of reactive oxygen and nitrogen species in cellular iron metabolism. Free Radic Res 2006;40:263–72.
- Benzi G, Moretti A. Are reactive oxygen species involved in Alzheimer's disease? Neurobiol Aging 1995;16:661–74.
- Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 2008;4:278–86.
- D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 2007;8:813–24.
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13.
- Meneshian A, Bulkley GB. The physiology of endothelial xanthine oxidase: from urate catabolism to reperfusion injury to inflammatory signal transduction. Microcirculation 2002;9:161–75.
- Olsen LF, Hauser MJB, Kummer U. Mechanism of protection of peroxidase activity by oscillatory dynamics. Eur J Biochem 2003;270:2796–804.
- Coleman JW. Nitric oxide in immunity and inflammation. Int Immunopharmacol 2001;1:1397–406.
- Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 2006;6:173–82.
- Brasen JC, Barington T, Olsen LF. On the mechanism of oscillations in neutrophils. Biophys Chem 2010;148:82–92.
- Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, et al. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res 2002;91:1160–7.
- Tochhawng L, Deng S, Pervaiz S, Yap CT. Redox regulation of cancer cell migration and invasion. Mitochondrion 2013;13:246–53.
- Dhar SK, St Clair DK. Manganese superoxide dismutase regulation and cancer. Free Radic Biol Med 2012;52:2209–22.
- Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta 2013;1830:3289–303.
- Rhee SG, Woo HA, Kil IS, Bae SH. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem 2012;287:4403–10.
- Pryor WA, Houk KN, Foote CS, Fukuto JM, Ignarro LJ, Squadrito GL, et al. Free radical biology and medicine: it's a gas, man! Am J Physiol Regul Integr Comp Physiol 2006;291:R491–511.
- Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem 1998;273:15366–72.
- Salmeen A, Andersen JN, Myers MP, Meng T-C, Hinks JA, Tonks NK, et al. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003;423:769–73.
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 2000;279:L1005–28.
- Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995;146:3–15.
- Kelekar A. Autophagy. Ann N Y Acad Sci 2005;1066:259–71.
- Hengartner MO. The biochemistry of apoptosis. Nature 2000;407:770–6.
- Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora's box opens. Nat Rev Mol Cell Biol 2001;2:67–71.
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2002;2:594–604.
- Wang X, Ryter SW, Dai C, Tang Z-L, Watkins SC, Yin X-M, et al. Necrotic cell death in response to oxidant stress involves the activation of the apoptogenic caspase-8/bid pathway. J Biol Chem 2003;278:29184–91.
- Ricci C, Pastuk V, Leonard J, Turrens J, Wilson G, Schaffer D, et al. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am J Physiol, Cell Physiol 2008;294:C413–22.
- Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced damage to DNA: mechanisms and measurement. Free Radic Biol Med 2002;32:1102–15.
- Zwacka RM, Dudus L, Epperly MW, Greenberger JS, Engelhardt JF. Redox gene therapy protects human IB-3 lung epithelial cells against ionizing radiation-induced apoptosis. Hum Gene Ther 1998;9:1381–6.
- Cossarizza A, Franceschi C, Monti D, Salvioli S, Bellesia E, Rivabene R, et al. Protective effect of N-acetylcysteine in tumor necrosis factor-alpha-induced apoptosis in U937 cells: the role of mitochondria. Exp Cell Res 1995;220:232–40.
- Chiba T, Takahashi S, Sato N, Ishii S, Kikuchi K. Fas-mediated apoptosis is modulated by intracellular glutathione in human T cells. Eur J Immunol 1996;26:1164–9.
- Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang B-H, et al. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol 2008;180:3072–80.
- Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol 2013; doi:10.1007/s00204-013-1034-4.
- Joseph B, Marchetti P, Formstecher P, Kroemer G, Lewensohn R, Zhivotovsky B. Mitochondrial dysfunction is an essential step for killing of non-small cell lung carcinomas resistant to conventional treatment. Oncogene 2002;21:65–77.
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007;26:1749–60
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 2009;28:3015–26.
- Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009;16:1040–52.
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 2006;25:3900–11.
- Janjetovic K, Misirkic M, Vucicevic L, Harhaji L, Trajkovic V. Synergistic antiglioma action of hyperthermia and nitric oxide. Eur J Pharmacol 2008;583:1–10.
- Lee YJ, Shacter E. Oxidative stress inhibits apoptosis in human lymphoma cells. J Biol Chem 1999;274:19792–8.
- Shacter E, Williams JA, Hinson RM, Sentürker S, Lee YJ. Oxidative stress interferes with cancer chemotherapy: inhibition of lymphoma cell apoptosis and phagocytosis. Blood 2000;96:307–13.
- Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep 2002;3:420–5.
- Bell EL, Klimova TA, Eisenbart J, Schumacker PT, Chandel NS. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol Cell Biol 2007;27:5737–45.
- Van Driel BE, Lyon H, Hoogenraad DCJ, Anten S, Hansen U, Van Noorden CJF. Expression of CuZn- and Mn-superoxide dismutase in human colorectal neoplasms. Free Radic Biol Med 1997;23:435–44.
- Cullen JJ, Mitros FA, Oberley LW. Expression of antioxidant enzymes in diseases of the human pancreas: another link between chronic pancreatitis and pancreatic cancer. Pancreas 2003;26:23–7.
- Senthil K, Aranganathan S, Nalini N. Evidence of oxidative stress in the circulation of ovarian cancer patients. Clin Chim Acta 2004;339:27–32.
- Hu Y, Rosen DG, Zhou Y, Feng L, Yang G, Liu J, et al. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J Biol Chem 2005;280:39485–92.
- Devi GS, Prasad MH, Saraswathi I, Raghu D, Rao DN, Reddy PP. Free radicals antioxidant enzymes and lipid peroxidation in different types of leukemias. Clin Chim Acta 2000;293:53–62.
- Qutub AA, Popel AS. Reactive oxygen species regulate hypoxia-inducible factor 1alpha differentially in cancer and ischemia. Mol Cell Biol 2008;28:5106–19.
- Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–82.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995;92:5510–4.
- Powell FL. Functional genomics and the comparative physiology of hypoxia. Annu Rev Physiol 2003;65:203–30.
- Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001;294:1337–40.
- Berra E, Ginouvès A, Pouysségur J. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep 2006;7:41–5.
- Acker T, Fandrey J, Acker H. The good, the bad and the ugly in oxygen-sensing: ROS, cytochromes and prolyl-hydroxylases. Cardiovasc Res 2006;71:195–207.
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001;292:464–8.
- Jung S-N, Yang WK, Kim JK, Kim HS, Kim EJ, Yun H, et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008;29:713–21.
- Schlegel HG. Aeration without air: oxygen supply by hydrogen peroxide. Biotechnol Bioeng 1977;19:413–24.
- Gottlieb E, Tomlinson IPM. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer 2005;5:857–66.
- Kozhukhar AV, Yasinska IM, Sumbayev VV. Nitric oxide inhibits HIF-1alpha protein accumulation under hypoxic conditions: implication of 2-oxoglutarate and iron. Biochimie 2006;88:411–8.
- Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol 2007;27:912–25.
- Chandel NS, Budinger GRS. The cellular basis for diverse responses to oxygen. Free Radic Biol Med 2007;42:165–74.
- Dirmeier R, O'Brien KM, Engle M, Dodd A, Spears E, Poyton RO. Exposure of yeast cells to anoxia induces transient oxidative stress. Implications for the induction of hypoxic genes. J Biol Chem 2002;277:34773–84.
- Grishko V, Solomon M, Breit JF, Killilea DW, LeDoux SP, Wilson GL, et al. Hypoxia promotes oxidative base modifications in the pulmonary artery endothelial cell VEGF gene. FASEB J 2001;15:1267–9.
- Hoffman DL, Brookes PS. Oxygen sensitivity of mitochondrial reactive oxygen species generation depends on metabolic conditions. J Biol Chem 2009;284:16236–45.
- Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, et al. Superoxide flashes in single mitochondria. Cell 2008;134:279–90.
- Chua YL, Dufour E, Dassa EP, Rustin P, Jacobs HT, Taylor CT, et al. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J Biol Chem 2010;285:31277–84.
- Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Volplo-Pulkki L-M. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res 2000;60:1789–92.
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 2004;56:185–229.
- Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer 1973;32:302–14.
- Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med 1998;339:900–5.
- Ichihara S. The pathological roles of environmental and redox stresses in cardiovascular diseases. Environ Health Prev Med 2013;18:177–84.
- Rhee SG, Chang T-S, Jeong W, Kang D. Methods for detection and measurement of hydrogen peroxide inside and outside of cells. Mol Cells 2010;29:539–49.