
Abstract
Sepsis is a clinical syndrome characterized by systemic inflammation, usually in response to infection. The signs and symptoms are very similar to Systemic Inflammatory Response Syndrome (SIRS), which typically occur consequent to trauma and auto-immune diseases. Common treatments of sepsis include administration of antibiotics and oxygen. Oxygen is administered due to ischemia in tissues, which results in the production of free radicals. Poor utilization of oxygen by the mitochondrial electron transport chain can increase oxidative stress during ischemia and exacerbate the severity and outcome in septic patients. This course of treatment virtually mimics the conditions seen in ischemia–reperfusion disorders. Therefore, this review proposes that the mechanism of free radical production seen in sepsis and SIRS is identical to the oxidative stress seen in ischemia–reperfusion injury. Specifically, this is due to a biochemical mechanism within the mitochondria where the oxidation of succinate to fumarate by succinate dehydrogenase (complex II) is reversed in sepsis (hypoxia), leading to succinate accumulation. Oxygen administration (equivalent to reperfusion) rapidly oxidizes the accumulated succinate, leading to the generation of large amounts of superoxide radical and other free radical species. Organ damage possibly leading to multi-organ failure could result from this oxidative burst seen in sepsis and SIRS. Accordingly, we postulate that temporal administration with anti-oxidants targeting the mitochondria and/or succinate dehydrogenase inhibitors could be beneficial in sepsis and SIRS patients.
Introduction
Sepsis is a clinical syndrome seen subsequent to severe infection. It has many of the same signs and symptoms, if not the same etiology, as Systemic Inflammatory Response Syndrome (SIRS). SIRS occurs following non-infectious inflammation (such as auto-immune diseases) as well as post-trauma (such as severe burns, surgery, etc.). Clinical definitions include high or low body temperatures, tachycardia, tachypnea, altered mental status, significant edema, and hyperglycemia. In general, high or low white blood cell counts are encountered as well as arterial hypotension. High levels of lactic acid are measured in blood, reflecting hypoperfusion and increased anaerobic metabolism in tissues.Citation1 As sepsis or SIRS worsen, multiple organs are involved and progress toward multi-organ failure (MOF).
For treatment of sepsis, rapid use of antibiotics and correction of physiologic perturbations are the common courses of action. Respiration is stabilized by administering oxygen. Tracheal intubation and mechanical ventilation are often required to maintain airway integrity. When hypotension is present, vasopressors are usually administered. Hypovolemia is countered by fluid administration, usually with crystalloids or human serum albumin, and lactate levels are monitored to adequately assess tissue perfusion. Numerous clinical trials have measured the effect of the available treatments for sepsis, SIRS, and MOF resulting from pejoration of these conditions (‘septic shock’). Activated protein C (Xigris) gained FDA approval for treatment of sepsis but was not widely adopted. As a result, it was voluntarily withdrawn from the market in 2011 after the PROWESS trial showed no overall benefit.Citation2 This anti-coagulant is presumed to act by inhibiting disseminated intravascular coagulation, which may lead to MOF. Among other treatments that have failed in clinical trials are inhibitors of endotoxin (LPS), inhibitors of the production of pro-inflammatory mediators (tumor necrosis factor, interleukin-1, platelet-activating factor, nitric oxide, etc.), and many other compounds that demonstrated efficacy in animal models of sepsis, SIRS, and MOF. For a particularly cogent discussion documenting the failure of these treatments, see the review by Marshall.Citation3 It is clear that the heterogeneity of the disease, the different causative organisms, the diverse genetic background of the patients, and the timing of intervention all contribute to the failure of most of these treatments for sepsis. In the future, the hope is that triaging patients according to rational criteria or new biomarkers will perhaps lead to the successful treatment of septic patients by preventing many deaths based on specific subgroups.
In the last decade, a very important finding has been the discovery of the cholinergic anti-inflammatory pathway. This pathway, investigated mainly by the laboratory of Kevin Tracey, involves stimulation of the tenth cranial nerve which indirectly controls TNF-alpha secretion. This has been used to reduce pathology in mouse sepsis,Citation4 and in a murine model of inflammatory bowel disease,Citation5 but, up to now, has only been used in humans for the treatment of rheumatoid arthritis.Citation6
Given the enormous complexity of sepsis and SIRS, both with respect to etiology and the biochemical pathways known to play a role in both its induction and resolution, and given the dramatic failure of treatment in well over 100 clinical trials, it may seem hubristic to point out one area of sepsis and SIRS that has been known for many years but which has not yielded many therapeutic approaches. This area is grounded in a physiological paradox. Oxidative stress occurs when the production of free radicals during respiration overwhelms the anti-oxidant capacity of cells or tissues to inactivate these molecules before they react with critical cell components such as nucleic acids, lipids, and proteins. When oxygen is delivered to cells at normal or above normal levels, the free radical production goes up in parallel with the oxygen tension. At lower than normal levels of oxygen, free radical production first decreases but as cells become more and more hypoxic, there is a change in metabolism and a paradoxical increase in the production of reactive oxygen species.Citation7 During sepsis and SIRS, this is precisely what happens – tissues become hypoxic and simultaneously there is an increase in free radical production and oxidative stress. It is this paradox that we wish to more fully examine, and then speculate on whether there may be hidden treatment possibilities underlying this pathology.Citation7
Free radical generation in mitochondria
To a first approximation, the currency of energy metabolism is ATP. The high-energy phosphate bond in ATP is derived from electron transport across a number of protein molecules and associated cofactors embedded in the inner membrane of the mitochondria. This electron transport chain eventually transfers electrons to, and thereby reduces, oxygen-yielding energy along the way for ATP synthesis. In general, oxygen consumption is regulated by the energy requirements of the cell, but this is not a perfect system. Free radicals such as nitric oxide (NO•) generated during electron transport can interact with nucleic acids, lipids, and proteins to produce toxic molecules like peroxynitrite. Normal cells have sufficient quantities of molecules like reduced glutathione, reduced thioredoxin, and other anti-oxidants to keep the level of free radicals low, and thus reduce damage to key cellular functions. However, during sepsis and SIRS, inflammation increases, resulting in dramatic changes in cell metabolism.
Accordingly, hypoxic tissues react by making new blood vessels that carry more oxygen to cells.Citation8 In addition, erythropoietin is produced causing the stimulation of red blood cell formation.Citation9 An increase in mRNAs coding for dozens to hundreds of proteins occurs when cells become hypoxic.Citation10 A major transcriptional regulator of these genes is the hypoxia-inducible factor (HIF).Citation11 In fact, there is an entire HIF family of genes. HIF is a heterodimeric protein composed of an alpha and beta subunit. The beta subunit, also known as aryl hydrocarbon receptor nuclear translocator, is constitutively transcribed and translated. The protein is stable and is found in cells under normal oxygen tensions. However, the alpha subunit is unstable under normal oxygen tension. It is degraded by the ubiquitin-proteasome system, and thus HIF does not function at normal oxygenation levels. HIF alpha degradation is inhibited when cells undergo extended exposure to hypoxic conditions. This allows for the stabilization of the HIF complex and its translocation to the nucleus to promote initiation of mRNA synthesis for multiple genes.Citation12
The oxygen dependent degradation by the proteasome was found to depend on enzymes called prolyl hydroxylases.Citation13 These enzymes hydroxylate highly conserved prolines in one segment of the HIF alpha subunit. This hydroxylation facilitates the interaction with the ubiquitin ligase (through the vonHippel-Lindau (vHL) protein) and thus proteasome degradation. These proline hydroxylases require alpha keto-glutarate and oxygen as substrates (with iron as a co-factor). During hypoxia, the prolyl hydroxylases are inactivated and the HIF alpha subunit is stabilized. Guzy and Schumacker have accumulated evidence that during hypoxic conditions seen in sepsis (as opposed to anoxic or nearly anoxic conditions), it is unlikely that the oxygen sensing mechanism is simply the lack of oxygen binding to its site on prolyl hydroxylases.Citation14 Rather, they postulate that the mitochondrial electron transport chain, particularly complex III, generates changes in free radical molecules and hydrogen peroxide in response to these hypoxic conditions. It is these changes that are sensed and lead to the stabilization of HIF alpha. The exact signaling mechanism that takes place has not yet been resolved.
How sepsis changes free radical generation and mitochondrial function
Sepsis and SIRS consist of a large inflammatory response, and a myriad of cytokines and cellular responses react to either the infection in sepsis or the trauma in SIRS.Citation15 This strong inflammatory response is counter-balanced by the synthesis of anti-inflammatory cytokines.Citation16 The exuberance of both the pro- and anti-inflammatory response has at times been postulated to be prognostic of either poor outcomes or protection from such outcomes.Citation17
Our focus here is the change in oxygenation and free radical production regardless of the inflammatory players involved. In sepsis and SIRS, there is decreased oxygen utilization in tissues, and this is observed both in model systems and in patients. Poor oxygen utilization leads to a decreased ratio of acetoacetate to beta-hydroxybutyrate with a continuing decrease in non-survivors and an increasing ratio toward normal levels (>0.7) in survivors.Citation18 Whether poor oxygen consumption is due to poor oxygen delivery to tissues or poor utilization by the mitochondrial electron transport chain has been the subject of some controversy. However, the end result is clear, namely that low levels of ATP are produced and more electrons leak from this chain, leading to increased free radical production and oxidative stress.Citation19 At least in sepsis, part of this oxidative burst may be triggered by activated macrophages, which can increase their expression of inducible nitric oxide synthase (iNOS). This leads to an increased production of nitric oxide, a known inhibitor of the mitochondrial electron transport chain.Citation19 Overall, there is a strong association between mitochondrial dysfunction and the severity and outcome of septic shock.Citation20
Targeting mitochondria with anti-oxidants
It has been well documented that sepsis/SIRS leads to the paradoxical augmentation of free radical oxygen and nitrogen species after hypoxia, resulting in the depletion of anti-oxidant reserves during this process. Therefore, it is logical to try to overcome the nefarious effects of this oxidative stress by increasing anti-oxidants in these patients. Moreover, such anti-oxidants often overcome sepsis and SIRS in a variety of animal models. A number of clinical trials using anti-oxidants have been completed, and, although they seem to aid in some of the physiological and biochemical abnormalities seen in sepsis, none have been FDA approved since the morbidity/mortality profile of patients remains unchanged.Citation3
In a randomized trial using the non-selective NOS inhibitor N-omega-methyl-L-arginine, an increase in mortality was observed, especially at higher doses, compared to placebo, even though some physiological functions improved.Citation21 It is possible that a selective inhibitor for iNOS might be a better choice for a clinical trial, since such a selective inhibitor, BYK191023, does prolong life in a sheep model compared to a norepinephrine control.Citation22 We shall return to the idea of targeting mitochondria for anti-oxidant therapy after discussing a recent advance in the field.
Recent finding on ischemia–reperfusion damage to mitochondria
Recently, a report by Chouchani et al.Citation23 on the mechanism of ischemia–reperfusion production of reactive oxygen species has led us to wonder about the applicability of these data to sepsis. The report starts with a comparative metabolomics study of hypoxic rodent tissues, including kidney, liver, heart, and brain. The conclusion is reached that the hypoxic conditions lead to an accumulation of mitochondrial succinate due to reversal of the succinate dehydrogenase enzyme acting on fumarate in the citric acid cycle. When reperfusion occurs, this accumulated succinate is rapidly reoxidized, generating large amounts of superoxide radical and other free radical species. Since the data for this interpretation are convincing, it leads us to wonder whether such a mechanism plays a similar role in sepsis.
Is sepsis treatment (oxygen and mechanical ventilation) after hypoxia creating massive conditions of ischemia–reperfusion in all affected organs?
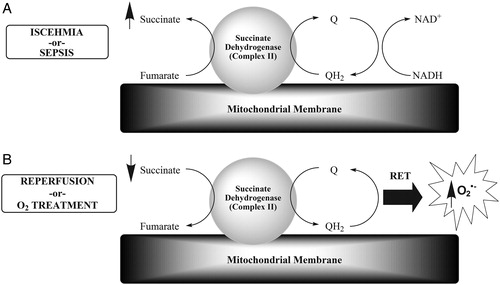
It is standard treatment for patients suffering from sepsis and SIRS to be administered oxygen, and this is often accompanied by mechanical ventilation.Citation24 These are exactly the conditions that mimic the ischemia (sepsis and SIRS, A) and reperfusion (oxygen treatment and mechanical ventilation, B) that lead to massive production of reactive oxygen species in the rodent model cited above. A priori, there is no reason to believe that sepsis as a source of ischemia–reperfusion should be any different than the methods used to induce ischemia–reperfusion in the Chouchani et al. study. On the contrary, there is already evidence that sepsis leads to accumulation of succinate.Citation25 This can be especially detrimental since succinate is a better candidate for oxidation than other citric acid cycle intermediates in the mitochondria of septic tissues.Citation26
Figure 1. This model is a simplified abstraction from Chouchani et al.Citation23 (A) The catalysis of fumarate to succinate in ischemia is initiated by succinate dehydrogenase (also known as complex II or succinate:ubiquinone oxidoreductase), resulting in the oxidation of dihydroubiquinone (QH2) to ubiquinone (Q), which is reduced back to QH2 by complex I via NADH/NAD+. Succinate dehydrogenase is the only enzyme in the tricarboxylic acid cycle that is embedded in the inner mitochondrial membrane, where it is also part of the electron transport chain. Our hypothesis is that sepsis/SIRS is equivalent to the induced ischemia in the study by Chouchani et al. (B) The accumulated succinate is then oxidized to fumarate by the same enzyme during reperfusion in the reverse manner. Our hypothesis is that oxygen (O2) treatment and mechanical ventilation are equivalent to reperfusion. This oxidation step is accompanied by superoxide radical (O2•−) production by reverse electron transport from complex I in the electron transport chain.
Can this be modulated and can treatment changes be made to test this possibility?
Understanding that at least some of the oxidative species damaging organs in sepsis may arise through rapid oxidation of succinate in mitochondria when oxygen therapy is started leads us to ponder whether this knowledge helps us re-think approaches to therapy in sepsis and SIRS. In the Chouchani et al.Citation23 study, intravenous injection of dimethyl malonate, a precursor of the succinate dehydrogenase inhibitor, malonate, decreased succinate production and subsequently protected brain injury in a mouse stroke model. Dimethyl malonate could be given to sepsis patients as a potential treatment when oxygen therapy is initiated. A comparison of the degree of organ injury versus a placebo group would be used to assess efficacy. The related compound, diethyl malonate, is actually a natural organic product found in grapes and strawberries. It is intriguing (but perhaps not related) that derivatives of malonate (specifically arylidene malonate) have been shown to inhibit TLR4 signaling after LPS stimulation.Citation27
There are many other anti-oxidant treatments that target mitochondria and could be used as adjuncts to oxygen therapy in these severe diseases. Lipophilic cations attached to anti-oxidants such as ubiquinone allow intra-mitochondrial accumulation of this anti-oxidant.Citation28 There are many other approaches to specifically channel anti-oxidants to the mitochondria reviewed by Galley.Citation29 Clinical trials for some of these, keeping in mind that the timing of treatment is paramount, might decrease the organ damage seen in severe sepsis and SIRS.
In conclusion, recent studies have revealed that many treatments which lead to tissue hypoxia induce a mitochondrial change in complex II, otherwise known as succinate dehydrogenase. This change reverses the succinate to fumarate reaction and reduces fumarate to succinate, which then accumulates in the mitochondria. Reperfusion leads to a burst of succinate to fumarate oxidation, resulting in the generation of many free radicals. This oxidative burst can eventually overwhelm the endogenous anti-oxidant supply thereby causing oxidative stress. We reason that sepsis may be considered such a source of hypoxia, and the immediate use of therapeutic oxygen in the hospital may be considered analogous to reperfusion. Thus, we postulate that oxygen administration in sepsis patients should be accompanied by mitochondrial redox reagents to eliminate the free radical burst associated with oxygen treatment.
Disclaimer statements
Contributors None.
Funding None.
Conflicts of interest None.
Ethics approval None.
References
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013;88(10):1127–40.
- Dolgin E. Trial failure prompts soul-searching for critical-care specialists. Nat Med 2012;18(7):1000.
- Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med 2014;20(4):195–203.
- Huston JM, Gallowitsch-Puerta M, Ochani M, Ochani K, Yuan R, Rosas-Ballina M, et al. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med 2007;35(12):2762–8.
- Ji H, Rabbi MF, Labis B, Pavlov VA, Tracey KJ, Ghia JE. Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol 2014;7(2):335–47.
- Koopman FA, Schuurman PR, Vervoordeldonk MJ, Tak PP. Vagus nerve stimulation: a new bioelectronics approach to treat rheumatoid arthritis? Best Pract Res Clin Rheumatol 2014;28(4):625–35.
- Bar-Or D, Bar-Or R, Rael LT, Brody EN. Oxidative stress in severe acute illness. Redox Biol 2015; 4C:340–5.
- Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, et al. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol 2000;156(3):965–76.
- Scholz H, Schurek HJ, Eckardt KU, Bauer C. Role of erythropoietin in adaptation to hypoxia. Experientia 1990;46(11–12):1197–201.
- Staudacher JJ, Naarmann-de Vries IS, Ujvari SJ, Klinger B, Kasim M, Benko E, et al. Hypoxia-induced gene expression results from selective mRNA partitioning to the endoplasmic reticulum. Nucleic Acids Res 2015. doi: 10.1093/nar/gkv167.
- Ruas JL, Poellinger L. Hypoxia-dependent activation of HIF into a transcriptional regulator. Semin Cell Dev Biol 2005;16(4–5):514–22.
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000;275(33):25130–8.
- Stolze IP, Mole DR, Ratcliffe PJ. Regulation of HIF: prolyl hydroxylases. Novartis Found Symp 2006;272:15–25; discussion-36.
- Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol 2006;91(5):807–19.
- de Jong HK, van der Poll T, Wiersinga WJ. The systemic pro-inflammatory response in sepsis. J Innate Immun 2010;2(5):422–30.
- Gomez HG, Gonzalez SM, Londono JM, Hoyos NA, Nino CD, Leon AL, et al. Immunological characterization of compensatory anti-inflammatory response syndrome in patients with severe sepsis: a longitudinal study*. Crit Care Med 2014;42(4):771–80.
- Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J Infect Dis 2000;181(1):176–80.
- Yassen KA, Galley HF, Lee A, Webster NR. Mitochondrial redox state in the critically ill. Br J Anaesth 1999;83(2):325–7.
- Brealey D, Singer M. Mitochondrial dysfunction in sepsis. Curr Infect Dis Rep 2003;5(5):365–71.
- Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002;360(9328):219–23.
- Lopez A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med 2004;32(1):21–30.
- Su F, Huang H, Akieda K, Occhipinti G, Donadello K, Piagnerelli M, et al. Effects of a selective iNOS inhibitor versus norepinephrine in the treatment of septic shock. Shock 2010;34(3):243–9.
- Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier S. Y, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014;515(7527):431–5.
- Luce JM. Pathogenesis and management of septic shock. Chest 1987;91(6):883–8.
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013;496(7444):238–42.
- Andrades ME, Morina A, Spasic S, Spasojevic I. Bench-to-bedside review: sepsis – from the redox point of view. Crit Care 2011;15(5):230.
- Zhang S, Cheng K, Wang X, Yin H. Selection, synthesis, and anti-inflammatory evaluation of the arylidene malonate derivatives as TLR4 signaling inhibitors. Bioorg Med Chem 2012;20(20):6073–9.
- Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci 2010;1201:96–103.
- Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth 2011;107(1):57–64.