
Abstract
Purpose:
The maraviroc (MVC) expanded access program (EAP) was initiated to increase MVC availability to patients with limited treatment options. Darunavir (DRV), raltegravir (RAL), and etravirine (ETV) were either recently approved or under regulatory review at study initiation and available for coadministration with MVC. Thus, the safety of MVC in combination with new antiretroviral therapies (ARVs) could be assessed. This open-label safety study of MVC was conducted at 262 sites worldwide in 1032 R5 HIV-positive treatment-experienced patients with limited/no therapeutic options.
Methods:
Study visits included screening, baseline, end of study or early discontinuation, and follow-up 30 days after last dose. Interim visits for HIV-1 RNA and CD4 cell counts occurred according to local HIV infection management guidelines. Safety data were analyzed overall and by subgroup based on ARV combination [MVC+optimized background therapy (OBT), MVC ± OBT+DRV/r, MVC ± OBT+RAL, MVC ± OBT+RAL+DRV/r, MVC ± OBT+RAL+ETV ± DRV/r].
Results:
Most (90.3%) adverse events (AEs) were of mild or moderate severity with few grade 3/4 events, discontinuations, or temporary discontinuations/dose reductions due to AEs or serious AEs. Similar results were observed across subgroups. Of treated patients, 79.9% and 50% had HIV-1 RNA < 400 copies/ml and < 50 copies/ml respectively, at the end of the study, early termination visits, or at last known status. Tropism changes and selection of MVC-resistant R5 virus, including high-level MVC dependence, were mechanisms of viral escape.
Conclusion:
MVC was well tolerated with virologic suppression observed in most patients.
Introduction
Maraviroc (MVC), a selective C–C chemokine receptor type-5 (CCR5) antagonist, is approved for use in treatment-experienced patients infected with CCR5-tropic human immunodeficiency virus subtype-1 (R5 HIV-1) at a dose of 300 mg twice daily (BID) or equivalent.Citation1,Citation2 Approval was based on data from two identically designed, registrational phase IIb/III studies, A4001027 and A4001028 (known as MOTIVATE-1 and -2, for Maraviroc Plus Optimized Therapy in Viremic Antiretroviral Treatment Experienced Patients), which were conducted in the USA and Canada (MOTIVATE-1) and in the USA, Canada, Australia, and Europe (MOTIVATE-2). In these studies, MVC 300 mg (or 150 mg in combination with potent CYP3A4 inhibitors) administered BID or once daily (QD) demonstrated safety and efficacy when used as part of combination antiretroviral (ARV) regimens. Both MVC regimens were significantly superior to placebo in all primary and secondary virologic and immunologic efficacy endpoints.Citation3–Citation5
A significant need remains for new drugs with novel mechanisms of action to be developed for treatment-experienced patients who have few or no remaining options due to multiclass drug resistance and/or intolerance to existing classes of ARV agents. Furthermore, several studies have demonstrated an association between the number of active drugs in the ARV regimen and virologic response rates.Citation4,Citation6–Citation8 It is therefore important that new drugs are combined with other active drugs in order to improve the likelihood of a sustained virologic response in heavily pretreated patients. In many cases, this would require the combination of two or more new drugs.
However, safety data are often limited for new drugs when used in a wider patient population. New combinations may result in unexpected side effects due to pharmacokinetic and/or pharmacodynamic interactions. Darunavir (DRV) was not permitted as part of optimized background therapy (OBT) in the MOTIVATE studies because it was not approved at the time the studies started and drug–drug interaction data were not available.Citation4,Citation6 Raltegravir (RAL) and etravirine (ETV) were both still in development at the time the MOTIVATE studies were conducted and were only approved for use in patients with HIV infection after MVC received its marketing authorization.
Following completion of the MOTIVATE studies and while MVC was under regulatory review, an expanded access program (EAP) (Study A4001050) was initiated in February 2007 to make MVC available to patients with limited or no approved treatment options due to resistance or intolerance and who required MVC to create a viable treatment regimen. The key objectives of this study were to allow for collection of safety data in a larger patient population more representative of those who now receive the drug in countries where MVC has been approved for use compared with the patients included in the phase IIb/III clinical study and to evaluate the safety of MVC in combination with new ARVs (DRV, RAL, and ETV) that were not available for use in the MOTIVATE studies. A secondary objective of the EAP was to evaluate the effectiveness of MVC in treatment-experienced patients who were followed according to local medical practice. In this paper we describe the safety and efficacy of MVC in the EAP, including its use in combination with the newer ARVs.
Methods
Study design, population, and conduct
Study A4001050 was an open-label, non-comparative, international, phase IIIb safety study of MVC in treatment-experienced patients with R5 HIV who had limited or no therapeutic options with approved therapies. The study was conducted at 262 centers throughout the world (Argentina, Australia, Austria, Belgium, Canada, Chile, Costa Rica, France, Greece, Hong Kong, India, Ireland, Italy, Malaysia, Mexico, Portugal, Puerto Rico, Romania, Spain, Switzerland, Taiwan, UK, the Netherlands, and the USA).
Patients were eligible for the study if they were at least 16 years of age (or minimum adult age as determined by local regulatory authorities or as dictated by local law); had limited or no approved treatment options available to them owing to resistance or intolerance; had failed to achieve adequate virologic suppression on their current regimen and had HIV-1 RNA ≥ 1000 copies/ml; and had only R5 HIV as verified by the original TrofileTM tropism assay (Monogram Biosciences, Inc., South San Francisco, CA, USA). Patients receiving other investigational ARV compounds through pre-approval EAPs or participation in Phase III or IV clinical studies were eligible for inclusion provided the two investigational agents offered the patient a regimen with two or three active ARVs (i.e. one approved treatment or no approved treatments were available to the patient due to resistance or intolerance), neither protocol prohibited the use of the other ARV, and the dosing of the two agents when used together was known. Patients receiving MVC or another CCR5 antagonist in an ongoing study that was being terminated or in which the patient's active dosing portion of the study was completed could be enrolled if the patient had only R5 HIV at the time of screening for the other study, had achieved a virologic response (>0.5 log10 reduction from baseline HIV-1 RNA concentration), and had never met the definition of virologic failure.
Key exclusion criteria were: prior treatment failure with any CCR5 antagonist; participation in any ongoing CCR5 antagonist study; or previous premature discontinuation in an MVC study. Patients were also excluded if they had a potentially life-threatening (grade 4) laboratory abnormality or medical condition (according to the Division of AIDS table for grading the severity of adult adverse events) that was still under investigation, unless a diagnosis had been established and was felt not to affect the risk/benefit assessment or eventual interpretation of safety results. The recommended MVC dose was 300 mg BID or equivalent, with dose adjustments according to the combination of drugs in the OBT or used for treatment of other intercurrent conditions.
Patients could withdraw from the study at any time at their own request or could be withdrawn at any time at the discretion of the investigator or sponsor. Additionally, patients were discontinued from the study if they met the definition of virologic failure:
| • | failing to achieve a reduction from baseline in HIV-1 RNA of ≥ 0.5 log10 copies/ml by the second viral load determination; | ||||
| • | experiencing a ≥ 0.5 log10 increase from nadir in HIV-1 RNA after achieving an HIV-1 RNA reduction from baseline of ≥ 0.5 log10 copies/ml; | ||||
| • | experiencing an HIV-1 RNA level of >1000 copies/ml after achieving an HIV-1 RNA level below the lower limit of quantification (LLOQ). | ||||
Study visits included screening, baseline, end of study or early discontinuation, and a follow-up visit 30 days after last dose of MVC as well as interim visits (suggested schedule: weeks 2–4, 8, 12, and every 12 weeks thereafter) where HIV-1 RNA and CD4 cell count testing were conducted. Patients were managed according to local guidelines for HIV infection and HIV-1 RNA and CD4 count testing were performed in local laboratories. Laboratory testing for safety was performed in a designated central laboratory (Covance, Indianapolis, IN, USA; Geneva, Switzerland; and Singapore). Coreceptor tropism was tested using the Trofile assay and a subset of patients with virologic failure was tested for MVC susceptibility (PhenoSense Entry; Monogram Biosciences), at screening or day 1 and end of study visits. The cut-off for MVC susceptibility was set at 95% maximal percent inhibition (MPI).Citation9
Safety was assessed by spontaneous reports, physical examinations, and laboratory test results in all patients who received at least one dose of study drug. Safety assessments were done at the screening, baseline, interim, and end of study or early discontinuation visits and included medical history (at screening only), complete or symptom-directed physical examination (at screening and end of study or early discontinuation visits only), vital signs and safety laboratory tests (e.g. hepatitis testing, pregnancy testing, hematology, and chemistry). Patients who met the following criteria were carefully evaluated:
| • | any patient (with normal baseline) who developed a grade 3 abnormality (with the exception of hypercholesterolemia; hypertriglyceridemia; asymptomatic creatine phosphokinase (CPK) elevations; aspartate aminotransferase (AST), or alanine aminotransferase (ALT) elevation in the absence of a total bilirubin elevation >2 times the upper limit of normal (ULN); or asymptomatic amylase or lipase elevations); | ||||
| • | any patient with abnormal baseline who developed a grade 3 abnormality and a level of ≥ 2 times baseline (with the exception of hypercholesterolemia; hypertriglyceridemia; asymptomatic CPK elevations; AST/ALT elevation in the absence of a total bilirubin elevation >2 times ULN; or asymptomatic amylase or lipase elevations); | ||||
| • | all patients who developed a grade 4 laboratory abnormality. | ||||
The study protocol was approved by the institutional review board or independent ethics committee at each study center. Written informed consent was obtained from all participants. The studies were performed in accordance with International Conference on Harmonisation Good Clinical Practice guidelines and applicable local regulatory requirements and laws. An independent data and safety monitoring board was responsible for oversight of the progress of the study, the study data, and safety considerations.
Data analysis
The safety analysis population included any patient who was allocated into one of the analysis groups and received at least one dose of study medication. Efficacy analyses were performed on data for all treated patients, regardless of whether they had a post-baseline visit. Patients from the EAP were pooled into three main groups for analysis:
| • | all EAP patients; | ||||||||||||||||||||||||||||
| • | EAP patients who received OBT that did not include novel drugs (DRV, RAL, ETV, or combinations of these) at baseline; | ||||||||||||||||||||||||||||
| • | EAP patients who received MVC with OBT containing a novel drug or combination of novel drugs as defined above at baseline; within this group, patients were subdivided according to which new drugs, or combinations of these, they were taking:
| ||||||||||||||||||||||||||||
All available data were included in the efficacy analyses. None of the efficacy analyses were based on the last observation carried forward. Endpoints included the following and were descriptive in nature:
| • | virologic response, determined by the proportions of patients with ≥ 0.5 log10 or ≥ 1.0 log10 reduction in HIV-1 RNA from baseline; | ||||
| • | proportions of patients who demonstrated a response to treatment in plasma HIV-1 RNA levels to < 400 copies/ml or < 50 copies/ml; | ||||
| • | change in CD4 count from baseline; | ||||
| • | proportions of patients with virologic failure and change in viral tropism from screening to end of study. | ||||
Results
Patient disposition, demographics, and baseline characteristics
Overall, 2584 patients were screened and 1047 patients assigned to MVC treatment. Of these, 1032 patients were treated with MVC BID. Overall, 916 patients (88.8%) completed the study and 116 patients (11.2%) discontinued (). The screening failure rate was 59.5% (1537/2584 patients), mainly due to the presence of CXCR4-using virus as determined by the original Trofile assay.
Table 1. Patient disposition overall and grouped by combination of novel ARVs
Of the 1032 patients assigned to treatment, 80.1% were male. Overall, 79.2% of patients were white, 10.3% were black, 5.2% were Asian, and 5.3% were of other race. Patients were aged between 16 and 80 years with a mean age of 45.6 years. The mean duration since first HIV diagnosis was 14.4 years (range: 0.3–27.0 years). The majority of patients (873; 84.6%) had R5 HIV recorded within the protocol-defined 42-day screening period. A further 2 patients had dual-mixed tropism recorded in this period, while 1 had a non-reportable tropism result. A further 156 patients had a screening tropism result recorded outside of the screening window. Of these patients, 151 had an R5 tropism result, 2 had dual-mixed tropism and 2 had a non-reportable tropism result. Therefore, a total of 1024 patients (99.2%) had an R5 tropism result recorded before study start. At baseline, mean HIV-1 RNA level was 4.3 log10 copies/ml and mean CD4 cell count was 259.6 cells/μl. Sixty patients (5.8%) tested positive for hepatitis B surface antigen and 158 patients (15.3%) were positive for hepatitis C antibody. A total of 1049 patients were included in the MOTIVATE studies of whom 414 received MVC QD, 426 received MVC BID, and 209 received placebo. Demographic characteristics were very similar to that of the EAP population; the mean baseline HIV-1 RNA level was 4.9 log10 copies/ml in the MVC QD, BID, and placebo groups. Mean baseline CD4 cell count [standard deviation (SD)] for patients in the combined MOTIVATE studies was 196.0 cells/μl (159.85) in the MVC QD group, 189.2 cells/μl (146.83) in the MVC BID group, and 186.4 cells/μl (133.52) in the placebo group.
In study A4001050, only 243/1032 (23.5%) of patients used MVC with an OBT that did not contain one of the newer ARVs. One hundred and fifty-eight (15.3%) included DRV as the only one of the newer drugs in their OBT, 155 (15.0%) RAL only, 263 (25.5%) DRV+RAL, and 213 (20.6%) ETV+RAL ( ± DRV).
Safety data
The median (range) duration of treatment was 174.0 (1–981) days (24.9 weeks) and was similar (169.0–181.0 days) for patients receiving each combination of novel ARVs. At least one all-causality AE was experienced by 70.4% of patients receiving MVC in the EAP. Overall, 2955 AEs were reported by 727 patients, of which 422 AEs experienced by 228 patients (22.1%) were considered related to the study drug. summarizes treatment-emergent adverse events by combination of novel ARV treatment groups. There were no clinically important differences by combination of novel ARV treatment(s) in the proportion of patients with AEs, the number of AEs, or the severity of AEs (). The most frequently reported all-causality AEs (>5% of patients) were diarrhea (9.3%), bronchitis (6.5%), headache (6.4%), nausea (5.9%), and pyrexia (5.7%). By comparison, the overall percentages of patients reporting AEs in the MOTIVATE studies were 90.6%, 92.3%, and 84.7% for the groups treated with MVC QD, MVC BID, and placebo groups, respectively. The most commonly reported AEs in the MOTIVATE studies were diarrhea and nausea.
Table 2. Summary of treatment-emergent AEs (all causality) by combination of novel ARVs
The majority of AEs (90.3%) were graded as mild or moderate in severity and there were few grade 3 or 4 events, discontinuations, or temporary discontinuations/dose reductions due to AEs (). The only grade 3 or 4 AEs that were reported by more than five patients were: hypertriglyceridemia (12 patients), pneumonia (9 patients), increased amylase (8 patients), increased gamma-glutamyl transferase (8 patients), chest pain (7 patients), increased lipase (7 patients), and nausea (6 patients).
Table 3. Treatment-emergent, treatment-related (maraviroc) serious adverse events — EAP
A total of 182 patients (17.6%) experienced serious adverse events (SAEs), including 16 deaths. Two of the patients who died had completed the study prior to death; one died 31 days after the last dose of MVC and the other 328 days after the last dose. For 26 patients, the SAEs (including three deaths) were considered to be related or possibly related to treatment with MVC, by either the investigator or the sponsor (). SAEs occurred at a similar frequency across the subgroups based on the combination of novel ARV drugs used (). The proportion of patients experiencing SAEs in the MOTIVATE studies was 15.0%, 16.9%, and 16.7% for MVC QD, MVC BID, and placebo, respectively.
Twenty-three patients experienced a total of 28 Category C AIDS-defining conditions (). One event was categorized as DAIDS (Division of AIDS) grade 1, 14 as grade 2, 8 as grade 3, and 4 as grade 4. The grade 4 Category C AIDS-defining conditions were: AIDS-related encephalopathy, cryptococcal meningitis, meningitis tuberculosis, and Mycobacterium kansasii infection. The only Category C AIDS-defining illnesses reported in more than two patients were esophageal candidiasis and pulmonary tuberculosis. There were no clinically important differences in the frequencies of Category C AIDS-defining illnesses when categorized by combination of novel antiretroviral treatment(s) (). In the MOTIVATE studies, 29 (7.0%), 22 (5.2%), and 16 (7.7%) patients in the MVC QD, MVC BID, and placebo treatment groups, respectively, had Category C AIDS-defining illnesses.
Table 4. Incidence of Category C AIDS-defining illnesses by combination of novel ARVs in the MVC EAP
Sixteen (1.6%) treatment-emergent non-AIDS-defining malignancies were reported. Only anal cancer (three patients), rectal cancer (three patients), basal cell carcinoma (two patients), and carcinoma in situ of penis (two patients) were reported in more than one patient (). There were no clinically relevant differences between the type and incidence of non-AIDS-defining malignancies between patients receiving each novel combination of ARV treatment(s). In the MOTIVATE studies, 11 (2.7%), 14 (3.3%), and 10 (4.8%) patients in the MVC QD, MVC BID, and placebo treatment groups, respectively, presented with treatment-emergent non-AIDS-defining malignancies.
Table 5. Incidence of treatment-emergent non-AIDS-defining malignancies in the MVC EAP
A total of 1014 patients receiving MVC in the EAP were evaluable for laboratory tests and no clinically significant median changes from baseline occurred in any laboratory test. The incidence of clinically significant grade 3 and 4 laboratory test abnormalities is summarized in . The incidence of grade 3 and 4 liver enzyme abnormalities was low and there were no cases of hepatotoxicity reported that met the criteria for Hy's Law.
Table 6. Incidence of maximum grade 3 and 4 laboratory test abnormalities (Division of AIDS) without regard to baseline abnormalities in the MVC expanded access program
Virologic and immunologic responses
There was a mean (SD) decrease from baseline in viral load by week 2 of 1.688 (0.8934) log10 copies/ml that was maintained to week 48 [(1.992 (1.1910) log10 copies/ml relative to baseline] and generally maintained up to the last assessment at week 144. At week 48, 89% (203/228) and 86% (197/228) of patients achieved >0.5 and >1 log10 reductions from baseline in HIV-1 RNA, respectively. Overall, 825/1032 (79.9%) and 516/1032 (50.0%) of treated patients achieved HIV-1 RNA < 400 copies/ml and < 50 copies/ml, respectively, at either end of study or early termination visits, or at last known status. The proportion of patients with viral load < 400 copies/ml increased from baseline [66/979 (6.7%) patients] to week 4 [562/773 (72.7%) patients] and was maintained to week 48 (184/228 [80.7%] patients). Similar results were observed for the < 50 copies/ml endpoint (). Mean (SD) CD4 cell count gradually increased from baseline [259.6 (186.29) cells/μl] to week 48 [393.7 (221.41) cells/μl].
Table 7. Proportions of patients with HIV-1 RNA < 400 and < 50 copies/ml at weeks 24 and 48
One hundred and ninety-two patients reached a virologic failure endpoint. The most common virologic failure endpoint (recorded in 44% of cases) was failing to achieve a reduction in HIV-1 RNA from baseline of ≥ 0.5 log10 copies/ml by the second viral load determination. Overall, 269 patients (26.1%) had reportable tropism results at the end of study assessment; 653 patients (63.3%) had non-reportable tropism results at the end of study assessment, reflecting the high proportion of patients with viral load < 400 copies/ml. Of the 227 patients with R5 HIV at screening and a valid tropism result at the end of study assessment, 71% (161/227) did not change tropism; the remainder changed from R5 to X4 (13 patients) or to dual mixed (53 patients).
Virologic failure and MVC susceptibility
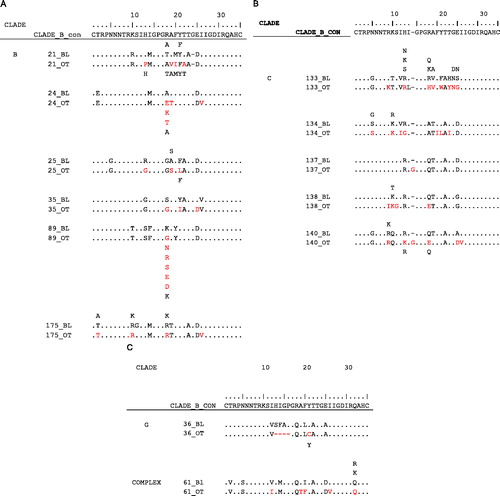
The evaluable virologic failure (eVF) population included those patients with protocol-defined virologic failure with plasma HIV-1 RNA >1000 copies/ml at the last on-treatment time point and at baseline (day 1). A total of 75 patients met these criteria. Notably, eVF patients tended to have low CD4 cell count and high plasma HIV-1 RNA levels at baseline compared with either the total virologic failure population or the total trial population (CD4 < 50 cells/mm3: 37.3%, 21.9%, 12.5%, respectively; plasma HIV-1 RNA ≥ 100,000 copies/ml: 34.7%, 28.1%, 22.7%, respectively). At day 1, 11 eVF patients (14.7%) had DM/X4 virus, 54 (73.0%) had R5 virus, and 10 (13.5%) did not have a tropism result. Of the 54 patients with R5 virus at day 1, 18 (33.3%) had a tropism that changed to DM/X4 on treatment; 14 (25.9%) had R5 virus resistant to MVC, 12 (22.2%) had R5 virus that remained susceptible to MVC, and 10 (18.5%) did not have a susceptibility result. Prolonged treatment on a failing regimen may have contributed to the development of resistance in a subgroup of patients (n = 6 treated for ≥ 365 days). The virus from one patient treated with MVC on a failing regimen for 538 days () showed full susceptibility to MVC at day 1 (), but was able to replicate efficiently only in the presence of MVC after treatment, as indicated by a highly negative MPI (–1230%; ). V3-loop amino acid sequence comparisons between day 1 and the last on-treatment time point were successfully completed with samples from 13 of the 14 patients. Although changes between the paired time points were observed for each sample, both the number and nature of the changes differed between patients and a consistent pattern of resistance was not observed (). This is consistent with observations from the MOTIVATE studies as well as the A4001026 (MERIT) study in treatment-naive patients, which demonstrated that although resistance was found to involve a common basic mechanism,Citation10 significant complexity of co-evolution at multiple sites, restricted commonality of sequence mutations, and divergent phenotypes prevent identification of any dependable pattern of mutations leading to resistance.Citation10–Citation12
Figure 1. Analysis of (A) plasma HIV-1 RNA over time; (B) baseline MVC susceptibility; and (C) on-treatment MVC susceptibility in patient with virus showing strong MVC dependence. Circles show the time points used for the baseline and on-treatment analysis of MVC susceptibility. MPI = maximal percent inhibition; MVC = maraviroc.
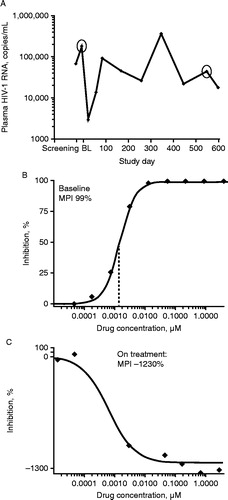
Figure 2. Insertions, deletions, and amino-acid changes between baseline and last on-treatment time point in the gp120 V3 loop of R5 virus from patients with evaluable virologic failure. Putative amino acids have been interpreted from the nucleotide sequence. Amino acids not seen in the baseline sequence are shown in red as are amino acids that have been selected from a mixture at baseline. The reference clade B sequence has been used for all three alignments. All amino acids that are identical to reference are shown with a dot. (A) Alignments of clade B sequences; (B) alignments of clade C sequences; (C) alignments of one pair of clade G and one pair of complex clade sequences.
Discussion
MVC demonstrated a favorable safety profile and was efficacious in the MOTIVATE studies.Citation3–Citation5 Consistent with data from other studies,Citation7,Citation8,Citation13,Citation14 subgroup analysis demonstrated that virologic response rates increased with the number of active drugs in the ARV regimen.Citation4,Citation6 Current guidelines recommend the use of at least two, and preferably three, fully active agents in a new regimen in patients with evidence of virologic resistance.Citation15,Citation16 In many heavily pre-treated patients, the combination of two or more new drugs would be required to increase the likelihood of a sustained virologic response.
Patients participating in the MVC EAP had the opportunity to receive a novel treatment regimen; that is, a combination of MVC and two or three new drugs (DRV, RAL, and ETV) that were either not commercially available, had only recently been approved or were under regulatory review at the time the study was initiated. More than 75% of patients in the EAP received at least one of the new ARVs as part of their OBT, and 46.1% received a combination of MVC plus at least two new ARVs. Therefore, and in spite of the limitations presented by the fact that this is an open-label study with no control group, the MVC EAP provided a unique opportunity for collection and analysis of safety data on new drug combinations that had not been evaluated previously in a treatment-experienced population.
Overall, MVC was well tolerated in this population with rates of AEs, grade 3 and 4 AEs, discontinuations due to AEs, and SAEs similar to or lower than those reported in the MOTIVATE studies. The incidence of Category C AIDS-defining events and non-AIDS-defining malignancies was very low. This, together with data from the 96-week follow-up of the MOTIVATE studiesCitation5 is reassuring in light of the lymphomas (and other cancers) reported in prior studies with another CCR5 antagonist.Citation17 Although individuals with the CCR5 delta 32 mutation are generally healthy, some immunological effects have been described,Citation18–Citation20 raising concern at the time regarding the long-term safety of CCR5 antagonists.
Despite concerns regarding the hepatic safety of CCR5 antagonists,Citation21,Citation22 an analysis of week 48 data from the MOTIVATE studies did not show a significant difference in severe hepatotoxic effects between MVC and placebo.Citation3 In the EAP, a very low rate of grade 3 and 4 liver enzyme abnormalities was recorded, confirming the hepatic safety of MVC in a wider HIV patient population. Additionally, a separate analysis of the subpopulation of EAP patients co-infected with hepatitis B or C demonstrated no increased incidence of AEs or severe liver enzyme abnormalities.Citation23
Comparison of the data from the EAP with that from the MOTIVATE studies demonstrated that MVC was equally well tolerated in this wider population and no additional safety concerns were highlighted. Finally, no significant differences were observed between the different subgroups based on the use of novel ARV combinations, demonstrating the safety of MVC as a component of each of the novel ARV regimens.
Patients eligible for the MVC EAP had very limited treatment options, but nonetheless the majority of patients (79.9%) achieved HIV-1 RNA < 400 copies/ml. A significant immunologic response was also observed, with an increase in mean CD4 count from 259.6 cells/μl at baseline to 393.7 cells/μl at week 48. This is similar to the virologic and immunologic responses observed in patients who had received two or more active drugs in their OBT regimen (based on overall susceptibility scores) in the MOTIVATE studies.Citation4,Citation6 Although the efficacy data from this open-label study should be interpreted with caution, the results indicate that even in a population of very treatment-experienced patients with limited treatment options remaining, the combination of MVC with one or more of the other new ARVs can elicit virologic suppression in the majority of patients.
The virologic findings in relation to mechanisms of viral escape were consistent with previous studies. As previously demonstrated, reduced maximal percent inhibition was the key observation of resistance to this inhibitor.Citation9 However, the data provide evidence not only of MVC resistance (i.e. the ability of the virus to replicate in the presence of MVC) but also of a rare case of MVC dependency (i.e. the need for MVC to be present for viral replication to occur efficiently) as indicated by negative values of percent inhibition in the presence of MVC seen in one patient. Altogether, these data further support the current hypothesis regarding the mechanism of resistance to CCR5 inhibitors.Citation9,Citation10
In summary, the data from the MVC EAP demonstrated that MVC was well tolerated in a population of patients managed according to local guidelines for the treatment of HIV, and in combination with other new ARVs to form novel combination regimens. Furthermore, the majority of patients participating in the EAP achieved an HIV-1 RNA level below the LLOQ, indicating the efficacy of MVC when used as a component of novel ARV combination regimens.
Acknowledgements
Monogram Biosciences provided the source data in . Marilyn Lewis provided assistance with sequence analysis for and drawing of Fig. 2.
Disclaimer Statements
Contributors
All authors were involved in study conception or design, or acquisition of data or analysis, and interpretation of data. All authors participated in revising the manuscript critically for important intellectual content, and approved the final version for publication.
Funding
This study was conducted by Pfizer Inc., and was sponsored by ViiV Healthcare. All listed authors meet the criteria set forth by the International Committee for Medical Journal Editors. Editorial support was provided by Complete Medical Communications Limited and MedThink SciCom and funded by ViiV Healthcare.
Conflicts of interest
Adriano Lazzarin has received honoraria and given expert testimony for ViiV Healthcare, Bristol-Myers Squibb, Janssen Pharmaceuticals, Gilead, Abbvie, and Merck Sharp & Dohme. Jacques Reynes has received payments for development of education materials and travel grants and has worked as a consultant on advisory boards for Pfizer Inc. and ViiV Healthcare. Jean-Michel Molina has served on advisory boards for Merck Sharp & Dohme, Gilead, Bristol-Myers Squibb, and Janssen Pharmaceuticals, and has received research grants from Merck Sharp & Dohme and Gilead. Srinivas Valluri owns stock and holds stock options in Pfizer Inc. Geoffrey Mukwaya and Elna van der Ryst own stock in Pfizer Inc. Jayvant Heera owns stock and holds stock options in Pfizer, Inc. Charles Craig has worked as a consultant for Pfizer Inc, and ViiV Healthcare and owns stock in GlaxoSmithKline. Juan G. Sierra-Madero has received grants from Pfizer Inc, and Merck Sharp & Dohme, speaker fees from Merck Sharp & Dohme, Bristol-Myers Squibb, Gilead, Janssen Pharmaceuticals, and ViiV Healthcare, and worked as a consultant for Merck Sharp & Dohme. Elna van der Ryst was an employee of Pfizer Inc during the time this study was performed.
Ethical approval
The study protocol was approved by the institutional review board or independent ethics committee at each study center. Written informed consent was obtained from all participants. The studies were performed in accordance with International Conference on Harmonisation Good Clinical Practice guidelines and applicable local regulatory requirements and laws. An independent data and safety monitoring board was responsible for oversight of the progress of the study, the study data, and safety considerations. This manuscript is an honest, accurate, and transparent account of the study being reported; no aspects of the study have been omitted, and any discrepancies from the study as planned have been explained.
References
- Selzentry [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2014.
- Celsentri [summary of product characteristics]. http://www.medicines.org.uk/emc/medicine/20386. Accessed May 19, 2014.
- Gulick RM, Lalezari J, Goodrich J, et al. MOTIVATE Study Teams. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med. 2008;359:1429–1441.
- Fätkenheuer G, Nelson M, Lazzarin A, et al. MOTIVATE 1 and MOTIVATE 2 Study Teams. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N Engl J Med. 2008;359:1442–1455.
- Hardy WD, Gulick RM, Mayer H, et al. Two-year safety and virologic efficacy of maraviroc in treatment-experienced patients with CCR5-tropic HIV-1 infection: 96-week combined analysis of MOTIVATE 1 and 2. J Acquir Immune Defic Syndr. 2010;55:558–564.
- Schapiro JM, Boucher CA, Kuritzkes DR, et al. Baseline CD4+ T- cell counts and weighted background susceptibility scores strongly predict response to maraviroc regimens in treatment-experienced patients. Antivir Ther. 2011;16:395–404.
- Clotet B, Bellos N, Molina JM, et al. POWER 1 and 2 study groups. Efficacy and safety of darunavir-ritonavir at week 48 in treatment-experienced patients with HIV-1 infection in POWER 1 and 2: a pooled subgroup analysis of data from two randomised trials. Lancet. 2007;369:1169–1178.
- Cooper DA, Steigbigel RT, Gatell JM, et al. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N Engl J Med. 2008;359:355–365.
- Westby M, Smith-Burchnell C, Mori J, et al. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol. 2007;81:2359–2371.
- Roche M, Salimi H, Duncan R, et al. A common mechanism of clinical HIV-1 resistance to the CCR5 antagonist maraviroc despite divergent resistance levels and lack of common gp120 resistance mutations. Retrovirology. 2013;10:43.
- Feyertag F, Xiang X, Lewis M, et al. Population dynamics of R5-tropic HIV-1 associated with maraviroc resistance. Antivir Ther. 2013;18:A34.
- Swenson LC, Chui CK, Brumme CJ, et al. Genotypic analysis of the V3 region of HIV from virologic nonresponders to maraviroc-containing regimens reveals distinct patterns of failure. Antimicrob Agents Chemother. 2013;57:6122–6130.
- Haubrich R, Cahn P, Grinsztejn B, et al. DUET-1: Week 48 results of a Phase III randomized double-blind trial to evaluate the efficacy and safety of etravirine (ETR; TMC125) versus placebo in 612 treatment-experienced HIV-1-infected patients. Poster presented at: 15th Conference on Retroviruses and Opportunistic Infections; February 3–6, 2008; Boston, MA, USA.
- Johnson MA, Campbell T, Clotet B, et al. DUET-2 study group. DUET-2: Week 48 results of a Phase III randomized double-blind trial to evaluate the efficacy and safety of etravirine (ETR; TMC125) versus placebo in 591 treatment-experienced HIV-1-infected patients. Poster presented at: 15th Conference on Retroviruses and Opportunistic Infections; February 3–6, 2008; Boston, MA.
- Thompson MA, Aberg JA, Cahn P, et al. International AIDS Society-USA. Antiretroviral treatment of adult HIV infection: 2010 recommendations of the International AIDS Society-USA panel. JAMA. 2010;304:321–333.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in HIV-1-infected Adults and Adolescents. 11-3-2013; Developed by the DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents; Convened by the Department of Health and Human Services (DHHS): 1–139.
- Gulick RM, Su Z, Flexner C, et al. Phase 2 study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-infected, treatment-experienced patients: AIDS clinical trials group 5211. J Infect Dis. 2007;196:304–312.
- Garred P, Madsen HO, Petersen J, et al. CC chemokine receptor 5 polymorphism in rheumatoid arthritis. J Rheumatol. 1998;25:1462–1465.
- Glass WG, McDermott DH, Lim JK, et al. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med. 2006;203:35–40.
- Dean M, Jacobson LP, McFarlane G, et al. Reduced risk of AIDS lymphoma in individuals heterozygous for the CCR5-delta32 mutation. Cancer Res. 1999;59:3561–3564.
- Moreno C, Gustot T, Nicaise C, et al. CCR5 deficiency exacerbates T-cell-mediated hepatitis in mice. Hepatology. 2005;42:854–862.
- Nichols WG, Steel HM, Bonny T, et al. Hepatotoxicity observed in clinical trials of aplaviroc (GW873140). Antimicrob Agents Chemother. 2008;52:858–865.
- Lazzarin A, Than S, Valluri SR, Heera J, Mukwaya G. Safety profile of maraviroc in patients coinfected with HIV-1 and hepatitis B or C included in the maraviroc expanded access program. HIV Clin Trials. 2012;13:83–89.