
Abstract
Background:
One of the more clinically relevant co-morbidities in HIV-infected patients is the development of progressive liver disease due to hepatitis B virus (HBV) or hepatitis C virus (HCV). In addition, hepatotoxicity has been observed with prolonged use of antiretroviral agents.
Objective:
To evaluate the hepatic safety of maraviroc in combination with other antiretroviral agents in HIV-1-infected subjects co-infected with HCV and/or HBV.
Methods:
In this 148-week randomized, double-blind, placebo-controlled, multicentre study (NCT01327547), subjects received maraviroc twice daily (n = 70) or placebo (n = 67) in combination with other antiretroviral agents. Primary endpoint: the percentage at week 48 of subjects with Grade 3 and Grade 4 ALT abnormalities, defined as >5 × upper limit of normal (ULN) if baseline ALT ≤ ULN or >3.5 × baseline if baseline ALT>ULN in the maraviroc versus the placebo arm.
Results:
At week 48, one subject in each group had met the primary endpoint definition. No subjects met protocol-defined liver stopping criteria and there were no cases of Hy's law or treatment-related hepatobiliary serious adverse events. No significant difference in change from baseline in enhanced liver fibrosis or hepatic elastography was observed between groups. Treatment-related hepatobiliary adverse events were reported in one and two subjects receiving maraviroc and placebo, respectively; discontinuations due to treatment-related AEs occurred in four and two subjects receiving maraviroc and placebo, respectively; two deaths were reported in the placebo group.
Conclusions:
The use of maraviroc does not increase hepatotoxicity in HIV-1-infected subjects co-infected with HCV and/or HBV through 48 weeks of treatment.
Introduction
As combination antiretroviral therapy (cART) has become more effective, progressive liver disease due to co-infection with hepatitis C virus (HCV) and/or hepatitis B virus (HBV) has emerged as an important cause of morbidity and mortality in HIV-1-infected subjects.Citation1,Citation2 Due to shared modes of transmission, co-infection with HIV-1 and HCV and/or HBV is common.Citation3 More importantly, HIV-1 has been shown to accelerate the course of HCV-associated liver disease as well as the progression of HBV disease, thereby clearly highlighting the need for treatment strategies in these specific subject populations.Citation4,Citation5
Maraviroc is a potent selective chemokine co-receptor type-5 (CCR5) antagonist that has been approved for the treatment of CCR5-tropic HIV-1 infection in both treatment-naive and treatment-experienced subjects in the United States,Citation6 as well as treatment-experienced subjects in the European Union.Citation7 Maraviroc is unique among approved antiretroviral agents in binding to a host receptor (CCR5) rather than a viral target. Maraviroc binding to CCR5 receptors prevents the interaction of HIV-1 gp120 with CCR5-tropic HIV-1 and thereby inhibits viral entry.Citation8–Citation11 Given the unique mode of action and use of a host cell target, initial concerns existed about the potential safety of maraviroc. Early development of other CCR5 antagonists had highlighted potential class-specific, long-term effects, particularly hepatotoxicity and malignancy. In addition, CCR5 deficiency had been reported to exacerbate T-cell-mediated hepatitis in animal models.Citation12 However, other studies reported reduced fibrogenesis in the presence of a CCR5 antagonist or in CCR5 knock-out mice.Citation13,Citation14 Further research measuring the effect of maraviroc on liver stiffness, when added to a current antiretroviral regimen, indicates a possible reduction in liver stiffness, as measured by hepatic elastography.Citation15
The purpose of this 148-week, randomized, double-blind, placebo-controlled, multicentre study, was to assess the hepatic safety of maraviroc in combination with other antiretroviral agents in subjects co-infected with HIV-1 and HCV and/or HBV.
Methods
Subjects
Key inclusion criteria
Eligible subjects were aged ≥ 18 years, HIV-1-positive on antiretroviral therapy (three to six drugs excluding ritonavir for ≥ 5 months prior to screening) with viral suppression (HIV-1 RNA < 50 copies/mL) for >3 months prior to screening, and co-infected with HCV and/or HBV as demonstrated by detectable HCV RNA and/or HBV surface antigen positivity at screening.
Key exclusion criteria
Key exclusion criteria included: alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) >5 × upper limit of normal (ULN), direct bilirubin >1.5 × ULN, prothrombin time prolonged >6 seconds, serum albumin < 2.8 g/dL, creatinine clearance ≤ 80 mL/min at screening; Child Pugh class C category (score >9); and underlying liver disease.
Study design and treatment
This is a 148-week, randomized, double-blind, placebo-controlled, multicentre study (ClinicalTrials.gov NCT01327547) conducted in compliance with the Declaration of Helsinki and all Good Clinical Practice Guidelines established by the International Conference on Harmonization. Subjects were enrolled at 38 different investigational centers located in the Czech Republic, France, Germany, Hungary, Poland, Spain, the United Kingdom, and the United States, including Puerto Rico. Central laboratory services were utilized and provided data included in the week 48 primary analysis. The study was supported by ViiV Healthcare.
Subjects were randomized 1:1 to receive maraviroc 150–600 mg twice daily (BID) or placebo for 144 weeks added to their concurrent antiretroviral regimen with a 4-week follow-up period at the end of the study. The maraviroc dose was selected based on the co-administration with potent inhibitors or inducers of CYP3A4/P-glycoprotein and reference was made to local prescribing information. Randomization was stratified according to HBV status, HIV protease inhibitor usage, and participation in a liver biopsy substudy. The sponsor was unblinded after the week 48 visit of the last enrolled subject while investigators and subjects remained blinded until week 144.
Concomitant medications
Trimethoprim sulfamethoxazole could only be continued if subjects were on stable therapy for ≥ 1 month. The following agents were not permitted during the study period: isoniazid, rifampin, rifapentine, rifabutin, and St. John's Wort; immunomodulators for the treatment of HIV-1 infection; or drugs that were contraindicated with any of the antiretroviral agents in the treatment regimen.
Study evaluations
Safety
The primary endpoint was the incidence of Grade 3 and Grade 4 ALT abnormalities, defined as >5 × ULN for subjects whose baseline ALT ≤ ULN, or >3.5 × baseline for subjects whose baseline ALT>ULN, through 48 weeks in the maraviroc versus the placebo arm.
Secondary endpoints included the time to development of Grade 3 and Grade 4 ALT abnormalities at week 48; incidence and time to development of Grade 3 and Grade 4 ALT abnormalities associated with change from baseline ALT >100 IU/L at week 48; percentage of subjects who met the criteria for the primary safety evaluation or who met the criteria for liver stopping rules at week 48; incidence of Hy's law abnormalities, defined as total bilirubin >2 × ULN with a simultaneous ALT or AST >3 × ULN, excluding subjects with an alkaline phosphatase >3 × ULN, at week 48; change from baseline in enhanced liver function (ELF) as measured by hyaluronic acid, tissue inhibitor of metalloproteinase (TIMP-1), and procollagen III N-terminal peptide (PIIINP) testing; change in baseline in hepatic elastography (only done at sites with access to Fibroscan™) at week 48; and the frequency and severity of AEs and laboratory abnormalities at week 48.
Efficacy
Secondary endpoints included the percentage of subjects with plasma HIV-1 RNA < 40 copies/mL at week 48; virus tropism, HIV susceptibility, and maraviroc resistance; change from baseline in plasma HCV RNA and HBV DNA at week 48; and change from baseline in CD4+ and CD8+ cell counts at week 48.
Sample size and statistical methods
Based upon the observed incidence of severe hepatotoxicity in two maraviroc Phase 3 treatment-experienced studies (A4001027 and A4001028), the anticipated event rate for this study was estimated to be 10% for the primary endpoint. A sample size of 60 subjects per group provided >70% to detect a clinically meaningful difference between the two treatment groups.
Analyses were conducted using the full analysis set population defined as a subject taking at least one dose of study drug. Primary endpoint was descriptively analyzed by comparing the proportion of subjects with Grade 3 and Grade 4 ALT abnormalities at week 48 in the two treatment groups of maraviroc and placebo. Total drug exposure in subject-years was calculated until time to event or end of study, whichever occurred first. Incidence rates per subject-years were also reported for the two treated groups. Kaplan–Meier analyses were performed to determine time to onset of the event.
Continuous endpoints such as ELF and hepatic elastography were analyzed using an analysis of covariance (ANCOVA) model with change from baseline as the response variable adjusting for treatment group, HBV status, PI-based regimen, and baseline measurement. Least-squares means (LSM) and 95% confidence intervals were reported. Missing values were imputed according to the last observation carried forward (LOCF) approach in all analyses conducted.
Results
Subject disposition and baseline demographic characteristics
A total of 218 subjects were screened for entry into the study, and of these, 137 subjects were randomized and received at least one dose of maraviroc BID (n = 70) or placebo (n = 67). The first subject visit occurred in May 2011, and the last subject visit for the week 48 analysis was in April 2013. Subjects will continue to be followed through week 148. Overall, 112 (81.8%) subjects were ongoing at the week 48 cut-off (). Baseline demographic characteristics were comparable between the treatment groups (, Supplementary Material 1). The majority of subjects were white (79.6%) and male (85.4%) with a mean (range) age of 48.3 (28.0–75.0) years. The mean (range) duration of HIV-1 infection was 14.4 (0–32.0) years. The most commonly used antiretroviral treatment was emtricitabine/tenofovir disoproxil fumarate in both the maraviroc and placebo groups (31.4 and 31.3%, respectively) (Supplementary Material 2). Ninety-four (68.6%) subjects were diagnosed with HIV-1 and HCV co-infection and 44 (32.1%) subjects were diagnosed with HIV-1 and HBV co-infection. Two subjects (1.5%) were co-infected with both HCV and HBV. Baseline total bilirubin, AST, and ALT values were graded as normal in 131 (95.6%), 108 (78.8%), and 100 (73.0%) subjects, respectively.
Figure 1. Subject disposition through week 48. AE, adverse event.
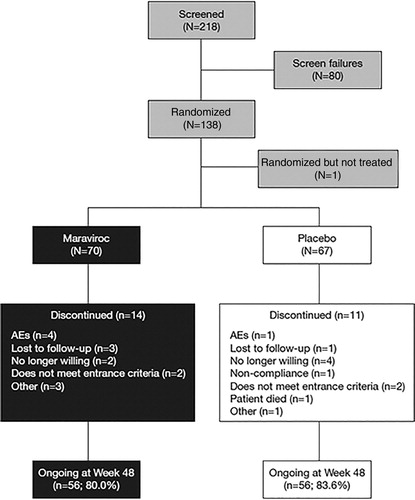
Table 1. Demographic and baseline characteristics
Safety
One subject in each treatment arm (1.4% maraviroc and 1.5% placebo) met the protocol-defined primary endpoint criteria of Grade 3 or 4 ALT abnormalities through 48 weeks. These subjects also had Grade 3/4 ALT abnormalities associated with changes from baseline ALT of >100 IU/L. Time to development of this primary safety endpoint in the subjects was 253 days (maraviroc) and 64 days (placebo). No subjects met the protocol-defined liver stopping criteria or Hy's law criteria through week 48.
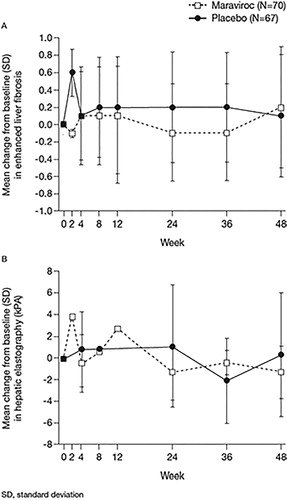
A total of 37 (17 maraviroc and 20 placebo) subjects had elastography performed at week 48; 53 subjects were included in the week 48 analysis using LOCF. At week 48, subjects showed a mean (SD) decrease from baseline in hepatic elastography of 1.3 kPa (2.41) and 0.3 kPa (5.71) on maraviroc and placebo, respectively. Subsequent ANCOVA analysis demonstrated that there is no statistically significant difference (P = 0.1457) between the treatment groups. There was also no statistically significant difference between the treatment groups in least-squares mean (LSM) change from baseline in enhanced liver function (ELF) test (P = 0.5201) ().
Figure 2. Change from baseline in (A) enhanced liver fibrosis and (B) hepatic elastography through week 48.
A summary of AEs is presented in . Thirty-eight treatment-emergent adverse events (TEAEs) in 18 (25.7%) subjects in the maraviroc group and 33 events in 19 (28.4%) subjects in the placebo group were considered to be related to treatment by the investigator. Of these, the most commonly reported events by system organ class were nervous system disorders (n = 6; 8.6%) and gastrointestinal disorders (n = 5; 7.1%) in the maraviroc group and gastrointestinal disorders in the placebo group (n = 11; 16.4%). Hepatobiliary TEAEs were reported in two (2.9%) subjects in the maraviroc arm [one subject was hospitalized due to cholangitis with hypertransaminasaemia (n = 1), and one subject had a TEAE of liver disorder (n = 1) reported as “hepatopathy due to interruption of tenofovir”] and four (6.0%) subjects in the placebo arm [cholestasis (n = 1); hepatic pain (reported as “liver pain on palpation”) (n = 1); hepatomegaly (n = 1); hyperbilirubinemia (n = 1)]. Of these, one event in the maraviroc arm [hypertransaminasaemia (n = 1)] and two events in the placebo arm [hepatomegaly (n = 1) and hyperbilirubinemia (n = 1)] were considered to be treatment-related. The majority of all treatment-related TEAEs in both groups were classified as Division of AIDS (DAIDS) Grade 1 or 2. Grade 3 treatment-related TEAEs were only reported in the maraviroc group and occurred in four (5.7%) subjects [abdominal discomfort (n = 1), increased ALT (n = 1), increased transaminases (n = 1), and ageusia (n = 1)]. No Grade 4 treatment-related TEAEs were reported in either group. Discontinuations due to treatment-related TEAEs occurred in four (5.7%) subjects in the maraviroc group compared with two (3.0%) subjects in the placebo group.
Table 2. Adverse event summary
Serious AEs (SAEs) were reported in 11 (15.7%) subjects in the maraviroc group and 12 (17.9%) subjects in the placebo group but were not considered to be treatment-related. No Category C AIDS-defining illnesses were reported in the maraviroc group; two (3.0%) subjects in the placebo group had Category C AIDS-defining illnesses (herpes simplex, pneumonia). No treatment-emergent malignancies were observed during the study.
Two deaths were reported in the placebo group during the study: a 36-year-old male died on Day 226 due to cardiac arrest, and a 63-year-old male died on Day 580 due to myocardial infarction.
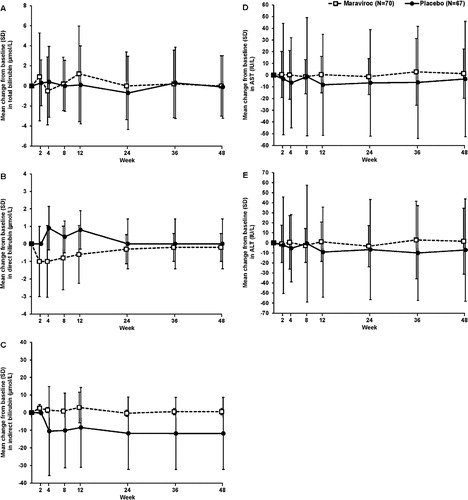
Minor changes from baseline were observed through week 48 for total bilirubin, direct bilirubin, indirect bilirubin, AST, and ALT (). The majority of laboratory abnormalities for liver function without regard to baseline were DAIDS Grade 1 or Grade 2. A shift summary of laboratory results by maximum DAIDS grade is presented in Supplementary Material 3. For both maraviroc and placebo, the percentage of subjects with normal total bilirubin, AST, and ALT at baseline who developed Grade 1 or Grade 2 total bilirubin was similar. One (1.4%) subject in the maraviroc group who had Grade 1 total bilirubin at baseline developed a maximum Grade 4 total bilirubin that was attributed to concomitant administration of atazanavir with normal direct bilirubin levels. Two subjects in the placebo group who had Grade 1 AST levels at baseline developed maximum Grade 3 AST abnormalities. One subject from each group, who had Grade 1 ALT at baseline, developed a maximum Grade 3 abnormality and subsequently also met the primary endpoint criteria.
Figure 3. Change from baseline in (A) total bilirubin, (B) direct bilirubin, (C) indirect bilirubin, (D) AST, and (E) ALT through week 48. ALT: alanine aminotransferase; AST: aspartate aminotransferase; SD: standard deviation.
Efficacy
There was a balanced rate of maintenance of HIV-1 RNA suppression ( < 40 copies/mL) at week 48 between the two treatment arms; 80.0% (n = 56) and 79.1% (n = 53) of subjects in the maraviroc and placebo groups, respectively. At week 48, one (1.4%) and three (4.5%) subjects in the maraviroc and placebo groups, respectively, had plasma HIV-1 RNA values >40 copies/mL. No tropism changes were observed in subjects with confirmed viral breakthrough and HIV-1 RNA >500 copies/mL. Analysis of covariance analysis at week 48 of the maraviroc and placebo groups showed HCV RNA LSM (SE) decreases of 3.22 (0.26) and 3.26 (0.25) IU/mL, and HBV DNA LSM (SE) decreases of 2.72 (0.32) and 2.87 (0.39) IU/mL, respectively. There was no statistically significant difference between the treatment groups in LSM change from baseline in HCV RNA (P = 0.8024) or HBV DNA (P = 0.7778) at week 48. The median change from baseline to week 48 in CD4+ count was 40.0 and 32.0 cells/μL for the maraviroc and placebo groups, respectively.
Discussion
Liver disease among HIV-1-infected subjects is a common and important cause of non-AIDS-related morbidity and mortality. Elevations in serum hepatic enzymes are common in subjects with HIV-1 and have been described in association with all major classes of antiretroviral therapy.Citation16 However, most liver disease among HIV-1-infected individuals is secondary to co-infection with HCV and/or HBV.Citation2 HIV-1 has been shown to accelerate the course of HCV-associated liver disease, as well as the progression of HBV disease, clearly underlining the need for treatment strategies in these specific patient populations.Citation4,Citation5,Citation17 Limited data from co-infected subjects in the maraviroc registrational program showed no evidence of increased hepatotoxicity.Citation18 We therefore investigated the hepatic safety of maraviroc in combination with other antiretroviral agents in subjects co-infected with HIV-1 and HCV and/or HBV.
Maraviroc was generally well-tolerated over 48 weeks of treatment in subjects co-infected with HCV and/or HBV. Results of the primary safety evaluation at week 48 showed that maraviroc did not increase hepatotoxicity in HIV-1-infected subjects compared with placebo. One patient in each treatment arm met the protocol-defined criteria of Grade 3 or Grade 4 ALT primary safety endpoint abnormalities. No subjects met the protocol-defined liver stopping criteria and there were no cases of Hy's law. There was no statistically significant difference between the treatment groups in the change from baseline in ELF test or hepatic elastography. While the potential of MVC to slow down fibrosis progression has been suggested previously,Citation13,Citation14 the relatively small sample size and short 48-week duration of follow-up on this study does not enable a complete assessment. Further follow-up of subjects in this study will continue as evaluation of the ELF test and hepatic elastography is incorporated as part of the defined secondary endpoints. Overall, the number of treatment-related TEAEs and the number of subjects reporting them were similar between the two groups. The relatively lower than expected hepatotoxicity in subjects co-infected with HIV-1 and HCV and/or HBV is supported by the inclusion of subjects who were already receiving virologically suppressive ARVs, thus removing the confounding effect of immune reconstitution. Furthermore, the relatively low hepatotoxicity rates are consistent with the findings of previous maraviroc studies.
There was no difference in viral suppression between the maraviroc and placebo groups through 48 weeks of treatment. There was no statistically significant difference in the change from baseline in HCV RNA or HBV DNA between the treatment groups, and no tropism changes were observed in subjects with confirmed viral breakthrough and HIV RNA >500 copies/mL.
The potential for hepatotoxicity with maraviroc has been previously studied. The results reported here are consistent with the findings of a previous study by Ayoub and colleagues, which investigated the hepatic safety of maraviroc across Pfizer-sponsored Phase 1–3 clinical trials.Citation18 These data together with data from clinical studies of the CCR5 antagonist vicriviroc, which showed no evidence of hepatotoxicity through 48 weeks of therapy, support our results that there is not a class effect among CCR5 antagonists.Citation19,Citation20 Furthermore, hepatotoxicity associated with the CCR5 antagonist aplaviroc that led to its discontinuation was considered likely to be unrelated to the mechanism of action (i.e. CCR5 inhibition) but rather due to the properties of the molecule itself.Citation21
In conclusion, the data reported here suggest that maraviroc does not increase hepatotoxicity in HIV-1-infected subjects co-infected with HCV and/or HBV. The 148-week follow-up in the context of ongoing pharmacovigilance activities will allow further assessment of the long-term hepatic safety of maraviroc.
Acknowledgments
Gerd Fätkenheuer would like to acknowledge the German Federal Ministry of Education and Research, who helped support her work on this study (research grant 01KG0915), and the support of the German Centre for Infection Research. Editorial support was provided by Karen Irving at Complete Medical Communications and Clint Smith at MedThink SciCom and was funded by ViiV Healthcare.
Disclaimer Statements
Contributors
All authors were involved in study conception or design, or acquisition of data or analysis, and interpretation of data. All authors participated in revising the manuscript critically for important intellectual content and approved the final version for publication.
Funding
This study was conducted by Pfizer Inc, and funded by ViiV Healthcare. JKR has received honoraria for consulting and/or speaking at educational events for Abbott Laboratories, AbbVie, Bionor, Bristol-Myers Squibb, Boehringer-Ingelheim, Gilead Sciences, Merck, Tibotec, and ViiV Healthcare. VS has received honoraria for consulting and/or speaking at educational events for Abbott, AbbVie, Boehringer-Ingelheim, Gilead Sciences, Janssen, Merck, Roche, and ViiV Healthcare. FP is an employee of and owns stock and stock options in Pfizer. GF has received grants and lecture fees from AbbVie, and has received lecture fees and served on advisory boards for Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, and Merck Sharp and Dohme. The employer of DMA, the University of California, has received funds from Pfizer in the form of grants and clinical trial support, including for this clinical trial. The employer of CBS, New York Medical College, received funding for the conduct of this study and other grants from ViiV Healthcare. CBS has been a clinical trial investigator for Merck, GlaxoSmithKline, Abbott Laboratories, Bristol-Myers Squibb, Pfizer, and OptiNose. GP has received research grants from Gilead Sciences and Bristol-Myers Squibb and payment for the development of educational materials and speaker honoraria from Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, and ViiV Healthcare. GM is an employee of and owns stock and stock options in Pfizer. SJ was an employee of Pfizer at the time of the study. JH owns stock and stock options in Pfizer. JAP has received consulting and lecture fees and research support from GlaxoSmithKline, Bristol-Myers Squibb, Abbott Laboratories, Janssen-Cilag, ViiV Healthcare, and Boehringer Ingelheim; consulting fees and research support from Pfizer; consulting and lecture fees from Gilead Sciences, Merck Sharp and Dohme and Janssen-Cilag; research support and lecture fees from Roche; and research support from Schering-Plough.
Conflicts of interest
All other authors report no conflicts of interest.
Ethics approval
This study was conducted in compliance with the Declaration of Helsinki and all Good Clinical Practice Guidelines established by the International Conference on Harmonisation. The final protocol, amendments, and informed consent documentation were reviewed and approved by the Institutional Review Board and the Independent Ethics Committee of the investigational centres. All subjects provided written, informed consent.
References
- Bica I, McGovern B, Dhar R, Stone D, McGowan K, Scheib R, et al. Increasing mortality due to end-stage liver disease in patients with human immunodeficiency virus infection. Clin Infect Dis. 2001;32(3):492–497.
- Weber R, Sabin CA, Friis-Møller N, Reiss P, El-Sadr WM, Kirk O, et al. Liver-related deaths in persons infected with the human immunodeficiency virus: the D:A:D study. Arch Intern Med. 2006;166(15):1632–1641.
- Koziel MJ, Peters MG. Viral hepatitis in HIV infection. N Engl J Med. 2007;356(14):1445–1454.
- Rockstroh JK. Management of hepatitis C/HIV coinfection. Curr Opin Infect Dis. 2006;19(1):8–13.
- Thio CL, Seaberg EC, Skolasky R Jr, Phair J, Visscher B, Muñoz A, et al. HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS). Lancet. 2002;360(9349):1921–1926.
- Food and Drug Administration. Selzentry: Highlights of Prescribing Information. 2007;. http://www.accessdata.fda.gov/drugsatfda_docs/label/2007/022128lbl.pdf. Accessed June 13, 2014.
- European Medicines Agency. Celsentri (maraviroc), European Public Assessment Report. 2014;. http://www.ema.europa.eu/ema/index.jsp?curl = pages/medicines/human/medicines/000811/human_med_000689.jsp&murl = menus/medicines/medicines.jsp&mid = WC0b01ac058001d124. Accessed December 14, 2011..
- Dragic T, Trkola A, Thompson DA, Cormier EG, Kajumo FA, Maxwell E, et al. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc Natl Acad Sci U S A. 2000;97(10):5639–5644.
- Tsamis F, Gavrilov S, Kajumo F, Seibert C, Kuhmann S, Ketas T, et al. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J Virol. 2003;77(9):5201–5208.
- Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67(4):1268–1282.
- Seibert C, Ying W, Gavrilov S, Tsamis F, Kuhmann SE, Palani A, et al. Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology. 2006;349(1):41–54.
- Moreno C, Gustot T, Nicaise C, Quertinmont E, Nagy N, Parmentier M, et al. CCR5 deficiency exacerbates T-cell-mediated hepatitis in mice. Hepatology. 2005;42(4):854–862.
- Marra F. Chemokines in liver inflammation and fibrosis. Front Biosci. 2002;7:d1899–d1914.
- Schwabe RF, Bataller R, Brenner DA. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am J Physiol Gastrointest Liver Physiol. 2003;285(5):G949–G958.
- Nasta P, Gatti F, Borghi F, Chiari E, Paderni A, Carosi G. Liver stiffness (LS) change in HIV-hepatitis C (HCV) coinfected patients treated with CCR5 inhibitor based antiretroviral therapy. Abstract presented at: 51st Interscience Conference on Antimicrobial Agents and Chemotherapy; September 17–20, 2011; Chicago, IL.
- Mankhatitham W, Lueangniyomkul A, Manosuthi W. Hepatotoxicity in patients co-infected with tuberculosis and HIV-1 while receiving non-nucleoside reverse transcriptase inhibitor-based antiretroviral therapy and rifampicin-containing anti-tuberculosis regimen. Southeast Asian J Trop Med Public Health. 2011;42(3):651–658.
- Macías J, Berenguer J, Japón MA, Girón JA, Rivero A, López-Cortés LF, et al. Fast fibrosis progression between repeated liver biopsies in patients coinfected with human immunodeficiency virus/hepatitis C virus. Hepatology. 2009;50(4):1056–1063.
- Ayoub A, Alston S, Goodrich J, Heera J, Hoepelman AI, Lalezari J, et al. Hepatic safety and tolerability in the maraviroc clinical development program. AIDS. 2010;24(17):2743–2750.
- Gulick RM, Su Z, Flexner C, Hughes MD, Skolnik PR, Wilkin TJ, et al. Phase 2 study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-infected, treatment-experienced patients: AIDS clinical trials group 5211. J Infect Dis. 2007;196(2):304–312.
- Zingman BS, Suleiman J, DeJesus E, Slim J, McCarthy M, Lee M, et al. Vicriviroc, a next generation CCR5 antagonist, exhibits potent, sustained suppression of viral replication in treatment-experienced adults: VICTOR-E1 48-week results. Abstract presented at: 15th Conference on Retroviruses and Opportunistic Infections; February 3–6, 2008; Boston, MA.
- Nichols WG, Steel HM, Bonny T, Adkison K, Curtis L, Millard J, et al. Hepatotoxicity observed in clinical trials of aplaviroc (GW873140). Antimicrob Agents Chemother. 2008;52(3):858–865.