Abstract
Objective
To determine the frequency of beta-thalassemia minor in subjects with no family history of hemoglobinopathy.
Methods
Subjects were self-recruited on thalassemia day by advertisement through media. Those with indexed cases of beta-thalassemia major were excluded. Participants were interviewed regarding their marital status and screening of partners. Complete blood counts and peripheral smear review were performed on EDTA samples. Hemoglobin (Hb) electrophoresis was performed in cases with mean corpuscular volume (MCV) <76 fl, mean corpuscular Hb (MCH) <27 pg. HbA2 level >3.5% was diagnostic for beta-thalassemia trait.
Results
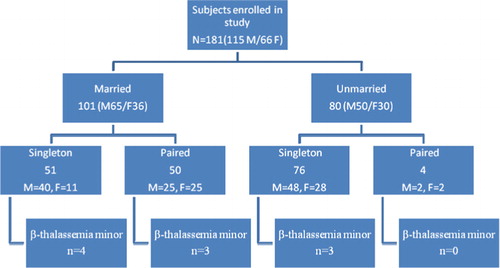
Out of 192 subjects, 11 were excluded based on family history of beta-thalassemia major and minor. Remaining 181 subjects (115 males and 66 females) were enrolled for further analysis. Median age was 27±9.7 years and included 101 married and 80 unmarried individuals. The mean Hb was 12.6 g/dl. MCV <76 fl and MCH <27 pg was seen in 29 subjects. Diagnosis of beta-thalassemia trait was made in 10 subjects (5.5%).
Conclusion
Though the carrier rate quoted is similar to previous studies, targeting families with indexed cases for screening might result in failure of carrier detection, since a large population would be overlooked. Implementation of national screening program is the need of the hour in Pakistan to evaluate the true burden of beta-thalassemia.
Introduction
Pakistan is home to over 170 million peopleCitation1 and the occurrence of hereditary hemoglobin (Hb) disorders in this population has been known for a very long time. Thalassemias are a heterogeneous group of hereditary Hb disorders with beta-thalassemia being the most common in our part of the world.Citation1 Despite several studies identifying beta-thalassemia mutations in Pakistan,Citation2–Citation4 the frequency of beta-thalassemia trait was reported two decades earlier based on Hb electrophoresis. This was reported as high as 6–13% in some of the major and populated cities like Lahore, Rawalpindi and Karachi.Citation5 It is estimated that 9.8 million carriers of beta-thalassemia existCitation6 and approximately 9000 neonates are born annually with beta-thalassemia major in Pakistan.Citation7 The average life expectancy for a patient with beta-thalassemia major is 10 years in our country with the current disease burden of 90 000–100 000 living patients.Citation8 There are several contributing factors responsible for this high mortality, including unavailability of safe blood,Citation9 decreased awareness of antenatal screening,Citation10 lack of education and poverty,Citation11 and poor overall health care system. The government of Pakistan has spent 0.6–1.1% of its GDP and 5.1–11.6% of its developmental expenditure on health over the last 10 years. The account of private sector health services is not included in this figure. The average yearly cost of regular blood transfusion therapy is $627 and that of iron chelation therapy would cost another $5100. Pakistan’s GDP in 2010 was reported as $161.99 billion and average household income ranges $1200–$1500/year.Citation12 These figures portray the economical burden on a family having one or more children with beta-thalassemia major. Recently, literacy rate has approached 55%,Citation13 but people having strong rural backgrounds and tradition of consanguineous marriages within their own caste are not uncommon. Lack of comprehensive awareness programs regarding inheritance of thalassemia has led to increased carrier marriages. Religious myths and misconceptions have prevented people from family planning and termination of pregnancies. Management of thalassemia major is difficult, especially for a resourced strained country like Pakistan; hence, prevention should be targeted for its eradication. One of the possible solutions would be to prevent the birth of thalassemic children by premarital carrier detection and or ante-natal screening.
Comprehensive beta-thalassemia carrier screening programs are prevalent worldwide.Citation14 In Pakistan, it is practiced in an ad hoc manner by identifying index cases in antenatal clinics. It is often based on the practitioner’s decision with no consistent screening policy other than requesting complete blood count early in a woman’s pregnancy. Screening strategies using extended family testing and cascade testing have been described.Citation15,Citation16 We hypothesized that carrier rate of beta-thalassemia is greater than previously reported figure of 6% and hence designed a study to identify beta-thalassemia trait in subjects through chromatography instead of electrophoresis. The present report aimed in determining the frequency of beta-thalassemia trait in subjects having no history of hemoglobinopathy in their relatives by using chromatography.
Subjects and Methods
Sample collection
The 8th May 2010 was marked by commemoration of Thalassemia Day at Fatimid Foundation, Karachi, Pakistan. The latter is a non-profitable organization with blood banking facilities and hematological services. The day was celebrated by complimentary mass screening for beta-thalassemia trait. Subjects were self-recruited and were attracted by advertisement through national media. All participants aged more than 18 years were interviewed regarding their marital status and screening of partners. Subjects who were known carriers or had an indexed case of thalassemia major or minor in the family were excluded from the study.
Complete blood counts and Hb variants analysis
Five milliliters of EDTA venous blood sample was collected from each enrolled individual for complete blood counts (Beckman CoulterTM automated hematology analyzer; Beckman Coulter, Miami, FL, USA). Peripheral smear was reviewed using Leishman stain by two individual microscopists. Hb electrophoresis (Inter lab by Scientific Instrument Division) was performed when mean corpuscular volume (MCV) was <76 fl, and or mean corpuscular Hb (MCH) <27 pg. A HbA2 level of >3.5% was considered diagnostic for beta-thalassemia trait. All such samples with high HbA2 were reconfirmed through Bio-Rad variant II high performance liquid chromatography analyzer (beta-thalassemia short program; Bio-Rad laboratories, Hercules, CA, USA).
Data handling
All the data were entered on SPSS version 16 (SPSS Inc., Chicago, IL, USA) for computing means, standard deviation (SD), and range of all descriptive variables.
Ethical concerns
Participants were enrolled after verbal consent. Subjects identified as iron deficient or with beta-thalassemia trait were informed through reports and written results of chromatography were issued to all the participants of the study. Further counseling as need be was provided on individual basis. The computerized data were recoded to maintain anonymity.
Results
Demographics
One hundred and ninety two subjects (males = 118, females = 74) visited Fatimid Foundation on Thalassemia Day for their complete blood counts. We excluded 11 subjects based on family history of beta-thalassemia major (10) and thalassemia minor (1). Remaining 181 subjects (115 males and 66 females) were enrolled for further analysis. They were mostly young with a median±SD age of 27±9.7 years and included both married (101) and unmarried (80) individuals. Their details are summarized in . Twenty one individuals (11.6%) were engaged at the time of enrolment in study.
Figure 1. Demographics of participants of the study (n = 181).
Hematological parameters
The mean±SD Hb was 13.4±1.6 g/dl (range: 2.8–16.9) in males and 11.2±1.3 g/dl (range: 7.3–15.0) in females. Reduced red cell parameters like MCV <76 fl and MCH <27 pg along with hypochromic microcytic erythrocytes and target cells were seen in 28 (15.4%) subjects. Additionally, one subject having borderline red cell indices had anisocytosis and hypochromia on blood film. Other parameters are shown in . All 29 samples were analyzed through Hb electrophoresis and high performance liquid chromatography. Their red cell parameters and chromatography results are detailed in showing a diagnosis of beta-thalassemia trait in 10 subjects (5.5%) while one individual had sickle cell trait ().
Figure 2. Analysis of 29 subjects for Hb variant analysis based on red cell indices.
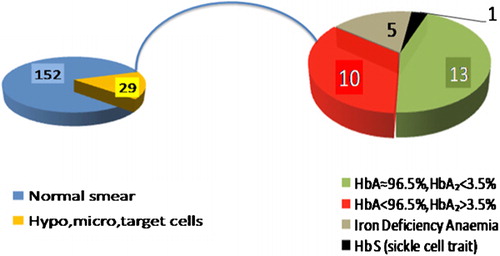
Table 1. Hematological parameters of n = 181 enrolled subjects (mean±SD)
Table 2. Hematological parameters with peripheral film findings in 29 subjects with hypochromic microcytic red cell indices (mean±SD)
Iron chemistry
Eighteen subjects having normal Hb electrophoresis and high performance liquid chromatography findings (HbA2 <3.5%) were tested for serum ferritin to rule out iron deficiency anemia. Serum ferritin was low in five subjects (two males and three females) confirming iron deficiency as the cause of hypochromic microcytic anemia. Authors believed that remaining 13 subjects having normal serum ferritin and HbA2 <3.5% represented either silent beta-thalassemia or alpha-thalassemia trait. Unfortunately, lack of resources limited us from further molecular analysis.
Discussion
The present study showed a prevalence rate of 5.5% for beta-thalassemia trait in general Pakistani population. The study underscored a number of important facts regarding thalassemia. It showed that presently awareness for thalassemia is high in big cities like Karachi where people were motivated for carrier detection. Also, it was mainly young people prior to their marriage (44% in our study) that showed inclination towards thalassemia testing.
Eight female carriers (4.4%) were identified from the antenatal group in our study. In Pakistan, screening is performed at antenatal clinics on the first visit after conceiving. Although the births of children with thalassemia major have fallen in various Mediterranean countries to between 10 and 50% of the 1970 rate, since diagnostic testing became available,Citation17 mandatory screening programs focusing mainly on premarital population implemented at national health policy level form an integral part of prevention strategy. In our study, the premarital group contained 1.6% (n = 3) carriers. World Health Organization guidelines published in 1998 prohibited compulsory genetic testing.Citation18 Nevertheless, thalassemia carrier screening has been included as part of an existing mandatory premarital blood test in Iran since 1991.Citation19 This form of mandatory premarital thalassemia screening also began in the Gaza Strip in 2000,Citation20 and in Saudi Arabia in 2003.Citation21 Carrier couples receive advice on the options available to them, one of which is cancelation of marriage, and they can then decide whether to marry each other. Apart from the ethical issues, such a program can be introduced and implemented at national legislative level in Pakistan to reduce the burden of thalassemia.
The strengths of our study include screening performed in subjects with no family history of hemoglobinopathy. Though the carrier rate is similar to what had been quoted in previous studies, targeting families with indexed cases for screening might result in failure of carrier detection strategies, since a large population would be overlooked for disease identification. Also, 2.7% of the subjects screened had iron deficiency and therefore carrier detection in these could have been missed due to falsely suppressed HbA2 levels.
Various countries have demonstrated the decrease in prevalence of beta-thalassemia trait by implementation of the national screening program which is the need of the hour in Pakistan. The true measure of a successful antenatal screening program is dependent on comprehensive education of community and medical professionals involved. Only then, will medical personnel have the adroitness to advise families appropriately so that couples can make informed reproductive choices and reduce the burden of a disease like thalassemia in Pakistan.
References
- prb.org [Internet]. Washington, DC: Population Reference Bureau; [Cited 2011 Apr 27]. Available from: www.prb.org/
- Khattak MF, Saleem M. Prevalence of heterozygous beta-thalassemia in northern areas of Pakistan. J Pak Med Assoc. 1992;42:32–4.
- Olivieri NF. The beta-thalassemias. N Engl J Med. 1999;341:99–109.
- Baig SM. Molecular diagnosis of beta-thalassemia by multiplex ARMS-PCR: a cost effective method for developing countries like Pakistan. Prenat Diagn. 2007;27:580–1.
- Baig SM, Azhar A, Hassan H, Baig JM, Kiyani A, Hameed U, et al.. Spectrum of beta-thalassemia mutations in various regions of Punjab and Islamabad, Pakistan: establishment of prenatal diagnosis. Haematologica. 2006;91:ELT02.
- Lodhi Y. Economics of thalassemia management in Pakistan. In: , Ahmed S, ed, editor. Thalassemia awareness week. Friends of Thalassemia; 2003.
- Ghani R, Manji MA, Ahmed N. Hemoglobinopathies among five major ethnic groups in Karachi, Pakistan. Southeast Asian J Trop Med Public Health. 2002;33:855–61.
- Rahman M, Lodhi Y. Prospects and future of conservative management of beta thalassemia major in a developing country. Pak J Med Sci. 2004;20:105–12.
- Riaz H, Riaz T, Ullah F, Aziz S, Khan MU, Pervaiz R, et al.. Assessment of the seroprevalence of viral hepatitis B, viral hepatitis C and HIV in multitransfused thalassaemia major patients in Karachi, Pakistan. Trop Doct. 2010;41:23–5.
- Ali SA, Donahue RM, Qureshi H, Vermund SH. Hepatitis B and hepatitis C in Pakistan: prevalence and risk factors. Int J Infect Dis. 2009;13:9–19.
- Bouhass RA, Kabouya EA, Smahi C, Benaceur SM, Aguercif M. Management of beta-thalassemias in a developing country. Experience of a pediatric service in Oran (Algeria). Ann Pediatr. 1992;39:115–9.
- Government of Pakistan, Statistics Division, Federal Bureau of Statistics Islamabad. Pakistan Social and Living Standards Measurement Survey (2004–05).
- unicef.org. UNICEF; [Cited 2010 Apr 20]. Available from: www.unicef.org/
- Trent RJ. Diagnosis of the haemoglobinopathies. Clin Biochem Rev. 2006;27:27–38.
- Baig SM, Din MA, Hassan H, Azhar A, Baig JM, Aslam M, et al.. Prevention of beta-thalassemia in a large Pakistani family through cascade testing. Community Genet. 2008;11:68–70.
- Dormandy E, Bryan S, Gulliford MC, Roberts TE, Ades AE, Calnan M, et al.. Antenatal screening for haemoglobinopathies in primary care: a cohort study and cluster randomised trial to inform a simulation model. The Screening for Haemoglobinopathies in First Trimester (SHIFT) trial. Health Technol Assess. 2010;14:1–160.
- Bain BJ. Screening of antenatal patients in a multiethnic community for beta thalassaemia trait. J Clin Pathol. 1988;41:481–5.
- Proposed international guidelines on ethical issues in medical genetics and genetic services (part I). Rev Derecho Genoma Hum. 1998;(8):219–23.
- Khorasani G, Kosaryan M, Vahidshahi K, Shakeri S, Nasehi MM. Results of the national program for prevention of beta-thalassemia major in the Iranian Province of Mazandaran. Hemoglobin. 2008;32:263–71.
- Tarazi I, Al Najjar E, Lulu N, Sirdah M. Obligatory premarital tests for beta-thalassaemia in the Gaza Strip: evaluation and recommendations. Int J Lab Hematol. 2007;29:111–8.
- Al-Suliman A. Prevalence of beta-thalassemia trait in premarital screening in Al-Hassa, Saudi Arabia. Ann Saudi Med. 2006;26:14–6.