Abstract
Acute promyelocytic leukemia (APL) is characterized by specific t(15;17), distinct morphologic picture, and clinical coagulopathy that contributes to the morbidity and mortality of the disease. This study was purposed to dissect the molecular mechanisms underlying telomerase-dependent arsenic trioxide (ATO)-induced cytotoxic and anti-proliferative effects in NB4 cells. ATO exposure was associated with transcriptional repression of Pin1, survivin, c-Myc, hTERT, and PinX1 along with an expressive enhancement in p73 mRNA level. Moreover, ATO treatment suppressed cell growth, viability and metabolic activity, exerted apoptosis, hindered telomerase activity, shortened telomere length, and dampened NF-κB activation. On aggregate, these issues indicate that ATO might preempt cell growth and proliferation in NB4 cells through suppression of Pin1-mediated NF-κB-dependent stimulation of telomerase and survivin.
Introduction
Acute promyelocytic leukemia (APL), a subtype of acute myeloid leukemia (AML), is characterized by t(15;17). This major type of cytogenetic abnormality in APL patients results in a reciprocal translocation which fuses the PML gene on chromosome 15 with the retinoic acid receptor alpha (RARα) gene on chromosome 17.Citation1 The PML-RARα protein has been thought to function as a repressor of retinoic acid (RA) signaling through recruiting the histone deacetylase complexes as well as DNA methyltransferase enzymes which cause methylation of RA-responsive promoters and transcriptionally repressing RARα-target genes.Citation2
Arsenic trioxide (ATO) has been used successfully in treatment of both newly diagnosed and relapsed patients with APL.Citation3 As a single agent, it induces complete remissions in vivo and triggers apoptotic death in APL cells.Citation4 An overwhelming number of studies imply that ATO induces apoptosis in a variety of tumor cells including AML,Citation5 multiple myeloma,Citation6 and neuroblastoma.Citation7 A substantial body of evidence indicates that ATO exerts variegated apoptotic-inductionary effects on tumor cells through caspase-3 activation,Citation8 modulation of the intracellular glutathione redox system and oxidative injuryCitation4 and induction of mitotic arrest due to inhibition of spindle apparatus and microtubuline formation.Citation5 In addition, there is ample evidence that ATO might suppress growth and proliferation of tumor cells through inhibition of telomerase and shortening of telomere length.Citation9 Although ATO has been widely used against APL, the molecular mechanisms underlying its anti-proliferative effects are not fully understood. In addition, the inhibitory mechanisms of ATO on human telomerase in tumor cells are less well understood.
New findings have underscored the pivotal roles of nuclear factor kappa B (NF-κB) signaling module in multistage tumorigenesis.Citation10 It has been suggested that NF-κB can govern all six hallmarks of cancer through controlling genes involved in proliferation, evasion from apoptosis, angiogenesis, invasion, and inflammation.Citation10 In closing, it has been suggested that NF-κB contributes to the unrestrained tumor cell proliferation through induction of both telomerase and survivin.Citation10 Given this, pharmacologically inhibition of NF-κB might promote programed cell death in tumor cells through repression of telomerase and survivin. An accumulating body of evidence indicates that survivin, the smallest member of the inhibitor of apoptosis family of proteins, antagonizes apoptosis,Citation11 promotes angiogenesis, and resistance to anti-cancer drugs.Citation12 Survivin has been implicated as a central player in multiple cellular networks including chromosomal passenger complex and the multilayered anti-apoptotic organization.Citation13 Given the nodal functions of survivin in multistage tumorigenesis and its cancer cell-specific expression, therapeutic disabling of survivin has produced massive interests in human hematological malignancies.
It has been shown that the prolyl isomerase Pin1 is up-regulated in many human cancers. It is thought Pin1 bind to the p65 subunit of the NF-κB module and this interaction is suggested to play critical roles in the promoter activity of the NF-κB signaling network.Citation14 In closing, a recent study by Atkinson et al.Citation15 has suggested that Pin1 is a positive regulator of p65. Selective targeting of Pin1 by specific short hairpin RNA prevents phosphorylation and activation of p65 and it is collectively indicated that Pin1 expression might lead to enhanced NF-κB signaling.Citation15 Given these results, we hypothesized that ATO-induced cytotoxic effect in NB4 leukemic cells may be modulated through suppression of NFkB-dependent induction of hTERT due to down-regulation of Pin1 transcription. To examine the probable mechanisms underlying telomerase-dependent ATO-induced cell death in APL, NB4 leukemic cells were subjected to treatment with various concentrations of ATO and succeeding cell viability index, growth kinetic, telomerase activity (TA), telomere length, NFkB phosphorylation, and the transcriptional alteration of Pin1, SVV, hTERT, p73, and other related target genes were investigated.
Materials and methods
Cell line and ATO treatment
Human APL cell line NB4 was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (Invitrogen, Gaithersburg, MD, USA), 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin G (Invitrogen), and 100 µg/ml streptomycin (Invitrogen). Cells were incubated at 37 °C in humidified atmosphere containing 5% CO2.
Assessment of cell growth and viability
The NB4 cells (2 × 105 /ml) were cultured in absence and presence of varying concentrations of ATO, ranging 0.25, 0.5, 0.75, 1, and 2 µM. Cell growth and viability were examined by Giemsa staining and trypan-blue dye exclusion, respectively. For detailed examination, cells were stained with 5 mg/ml Hoechst 33342 (Sigma, St Louis, MO, USA) at 37 °C for 30 minutes and apoptotic cells were identified using a fluoromicroscope. Alternatively, the proliferation of the NB4 cells during treatment with 0.5, 1 and 2 µM of ATO was measured using MTT (3-[4, 5-dimethylthiazol-2-yl-2, 5-dipenyl tetrazolium bromide) reagent (Sigma) in phosphate-buffered saline (PBS) at a final concentration of 0.5 mg/ml. After 4 hours of incubation at 37 °C, the formazan product was dissolved in 0.5N HCl, 5% triton x-100, and 45% 2-propanel (final concentrations). The intensity of dissolved formazan crystal was measured at 590 nm.
Assessment of cell apoptosis with fluorescence microscopy
The NB4 cells treated with 0.5, 1, and 2 µM of ATO were dual stained for 20 minutes with 10 µM each of Hoechst 33342 and propidium iodide (PI) and examined with a fluoromicroscope using red and blue filters. The captured images were processed using the Leica FISH imaging System (Leica, Milton Keynes, Buckinghamshire, UK). Percentage of cells stained with PI (red) was regarded as the percentage of late apoptotic cells.
Assessment of cell apoptosis with flow cytometry
About 1 × 106 of NB4 cells exposed to varying concentrations of arsenic for 48 hours were collected, washed twice with cold PBS, and fixed in 70% ethanol overnight. The cells then were incubated in 1 ml PBS containing 50 µg/ml PI and 1 mg/ml Annexin-V-PE. The fluorescence intensity of PI and Annexin-V were simultaneously determined by flow cytometry (BD Biosciences, Heidelberg, Germany).
Assessment of TA
TA in response to 0.5, 1, and 2 µM of ATO was determined by Telo TAGGG Telomerase PCR ELISA kit (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer's instructions. The polymerase chain reaction (PCR) amplified telomerase products obtained in these assays were also visualized on 8% polyacrylamide gel electrophoresis (PAGE) and the ladder was detected by staining with sliver nitrate (Sigma). The gel image was captured and analyzed using Quantity One and Multi-Analysis softwares (Bio-Rad Laboratories, Hercules, CA, USA). TA was calculated as the ratio of the intensity of telomerase ladders to the intensity of the 36-bp standard. Percentage of inhibition was calculated by comparing TA of arsenic-treated cells with TA of untreated cells. The results of study showed that input of 1000 NB4 cells extract with 30-cycle PCR amplification and 5 µl of PCR products for enzyme-linked immunosorbent assay (ELISA) detection and 10 µl of the PCR product for PAGE detection provided a quantitative and reproducible assay for TA in these cells.
Assessment of telomere length
Telomere length (terminal restriction fragment (TRF)) after exposure to 0.25, 0.5, and 0.75 µM of ATO was measured by using a non-radioactive chemiluminescent assay, using the Telo TAAGGG Telomere Length Assay kit (Roche Diagnostics) according to the manufacturer's instructions. Aliquots of 1.5 µg genomic DNA was digested with RsaI and HinfI and then resolved on a 0.8% agarose gel. The gel was depurinated, denatured, neutralized, and transferred to a positively charged nylon membrane (Roche Diagnostics). Filters were then prehybridized for 2 hours, hybridized with a digoxigenin-labeled telomeric probe at 65 °C for 16 hours and washed in 2x SSC/0.1% sodium dodecyl sulfate. Chemiluminescent detection was performed by using the anti-digoxigenin-AP Luminescent Detection kit (Roche Diagnostics), filters were incubated with a DIG-specific antibody covalently coupled to alkaline phosphatase, washed, and incubated with CDP-Star chemiluminescent substrate. Telomeric smears were visualized by exposing the membranes to a Lumi-Film Chemiluminescent Detection Film (Roche Diagnostics). The film image was captured and analyzed using Quantity One and Multi-Analysis softwares (Bio-Rad). Telomere peak values were measured by estimating the band size corresponding to the point with the highest signal intensity defining the highest concentration of telomere repeats. The peak value of TRF lengths were calculated and recorded as TLs.
Assessment of cell-based NF-κB phosphorylation by ELISA
The effect of ATO on NF-κB activation was surveyed by Cellular Activation of Signaling ELISA (CASE Kit, SABioscience, Frederick) as per manufacture's protocol. In brief, cells were plated onto 96-well plates at a density of 20,000 cell/well/100 µl. After treatment with 0.5, 1, and 2 µM of ATO at 37 °C for 48 hours, cells were fixed in a 8% formaldehyde:PBS solution and then stained with primary and secondary antibodies followed by exposure to developing and stop solutions provided with the kit. After normalizing with the relative cell number obtained by a cell staining buffer at 578 nm, the ratio of phosphorylated NF-κB to total NF-κB was obtained at 450 nm.
Assessment of gene expression by real-time quantitative PCR
FastPure RNA kit (Takara Bio, Inc., Otsu, Japan) was used to isolate total RNA from cultured cells. The quantity of RNA samples was assessed spectrophotometrically using Nanodrop ND-1000 (Nanodrop Technologies, Wilmington, DE, USA). Changes in mRNA expression of desired genes were surveyed by real-time PCR after reverse transcription of 1 µg RNA from each sample with PrimeScript RT reagent kit (Takara Bio) according to manufacture's specifications. Quantitative real-time reverse transcriptase-PCR was performed on a light cycler instrument (Roche Diagnostics, Mannheim, Germany) using SYBR Premix Ex Taq technology (Takara Bio). Ten microliters of SYBR Green master mix was added to 2 µl of cDNA samples, 0.5 µl of forward and reverse primers (10 pmol) in water plus 7 µl of nuclease-free water (Qiagen, Hilden, Germany) to conduct PCR in 20 µl reaction mixture. Thermal cycling conditions involved an initial activation step for 30 seconds at 95 °C followed by 45 cycles including a denaturation step for 5 seconds at 95 °C and a combined annealing/extension step for 20 seconds at 60 °C. Melting curve analysis was applied to validate whether all primers yield a single PCR product. Hypoxanthine phosphoribosyl transferase1 (HPRT1) was amplified as normalizer and fold change in expression of each target mRNA relative to HPRT1 was calculated based on 2−ΔΔct relative expression formulaCitation16. The nucleotide sequence of the primers used is given in .
Statistical analysis
Data are expressed as mean ± standard deviation. All experiments were performed in triplicate. For statistical analysis, the Student t test and one-way analysis of variance were applied. In order to compare between the control group and the experimental ones, Dunnett's multiple comparison tests was used. P values of <0.05 were considered significant.
Results
Effect of ATO on cell viability and proliferation
As shown in , ATO inhibited proliferation and viability of NB4 cell in dose- and time-dependent manners. A depicts the viability of NB4 cells after exposure to ATO which might be divided into two groups: the low concentrations of ATO (0.25–0.5 µM) in which the inhibitory effect of ATO is moderate and cells continue to proliferate at a reduced rate and the high concentrations of ATO (0.75–2 µM) in which the inhibitory rate of ATO is immediate after exposure. B and 1C show the suppressive effects of ATO on proliferation of NB4 cells using MTT assay. As shown by B, a dose-dependent reduction in proliferative rate of NB4 cells was observed. In addition, as shown by C, ATO exerted a time-dependent decrease in proliferation of NB4 cells.
Figure 1. Effect of ATO on proliferation and viability of NB4 cells. To quantify the effect of ATO on cell viability and growth, NB4 cells were grown in the presence of various concentrations of ATO. (A) Cell viability was thereafter assessed by trypan blue exclusion assay. Percent viability was normalized to the untreated cells. Data are shown as the mean ± SD from three independent experiments. Cell viability was also assessed by MTT assays. (B) A dose-dependent reduction in proliferative rate of NB4 cells. (C) A time-dependent decrease in proliferation of NB4 cells.
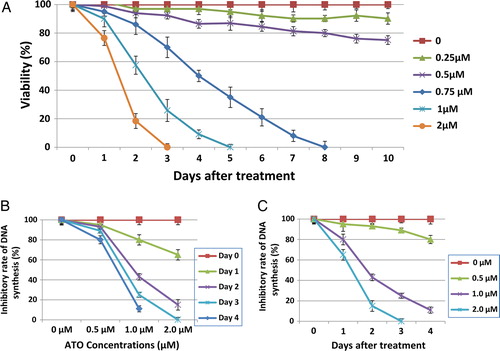
Induction of apoptosis by ATO
NB4 cells exposed to 0.5, 1, and 2 µM of arsenic were dual-stained with Hoechst 33342 and PI and analyzed by fluorescent microscopy. Hoechst can stain nucleus of all cells, while PI stains the cells with permeable membranes. This approach allows further distinction of late apoptotic/necrotic and living cells. As shown in A, images were captured by different filters set and processed using Leica FISH imaging system. The viable and apoptotic cell populations were quantitated. After 48 hours of incubation, there was a concentration-dependent (0.5–2.0 µM) increase in the rate of apoptotic cells (19–67%) compared with the untreated cells (<10%), whereas the viable cell population was reduced tremendously.
Figure 2. Induction of apoptosis by ATO. To determine whether ATO-induced cell death was due to apoptosis, the NB4 cells were cultured in the presence of various ATO concentrations for 2 days and thereafter dual stained with (A) Hoechst 33342 and PI dyes and examined by fluorescence microscope. The images were captured with red (for Hoechst) and blue (for PI) filters and merged using FISH imaging system. Percentage of cells stained with PI (red) was regarded as the percentage of apoptotic cells. (B) Annexin-V-PE and PI, early and late apoptosis was determined by flow cytometric analysis.
Flow cytometry analysis of apoptosis
ATO-treated NB4 cells were dual-stained with Annexin-V – PE and PI and analyzed by flow cytometry. Annexin V-PE-positive and PI-negative cells (annexin V+/PI−) were considered early apoptotic; the cells with both Annexin V-PE- and PI-positive (annexin V+/PI+) were considered late apoptotic/necrotic cells. After 48 hours of incubation, there was a dose-dependent (0.25–2.0 µM) increase in early and late apoptotic cells as shown in B. As concentrations of arsenic increased, significant increases of both early and late apoptotic cells were detected. At concentration of 2 µM, the NB4 cells underwent massive apoptosis; an average of 38% of annexin V+/PI− cells and 65% of annexin V+/PI+ cells were detected.
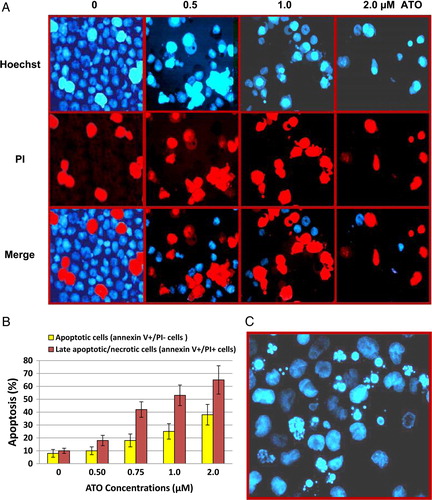
Inhibition of TA by ATO in NB4 cells
The effect of ATO treatment on TA was monitored by TRAP assay. The percent inhibition of TA determined by ELISA was correlated with the TRAP ladder detected by PAGE analysis. As shown in , ATO inhibited TA. Treatment of NB4 cells with low-dose arsenic resulted in a partial inhibition of TA. At higher concentrations of arsenic (1 and 2 µM), inhibition of TA started as early as 6 hours. TA was inhibited completely after 2 and 4 days treatment with 2 and 1 µM arsenic, respectively. The direct effect of arsenic on TA was also examined by adding different concentrations of ATO to the cell extract; the results showed no changes compared with their counter partners.
Figure 3. Inhibition of telomerase activity by ATO in NB4 cells. The NB4 cells were grown in presence of various concentrations of ATO. At the indicated time, cells were collected and TA was measured for 1000 cells equivalent per lane using PCR-based TRAP assay. (A) Analyzing of TRAP products of NB4 cells after treating with three doses of ATO (0.5, 1, and 2 µM). TRAP ladder was resolved on an 8% PAGE and visualized by silver staining. (B) NB4 cells treated with 1 µM ATO. (C) NB4 cells treated with 2 µM ATO. M, molecular marker (100 bP and 200 bP); 0 + , adding of arsenic to the cell extract.
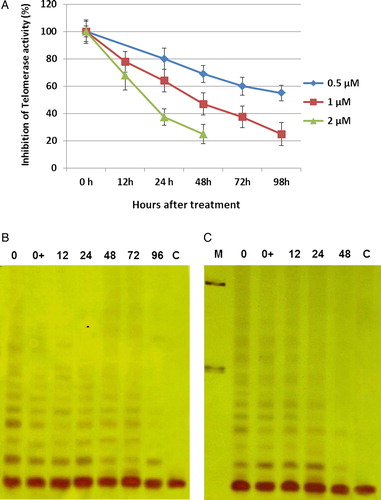
Telomere length shortening by ATO
To determine the effect of arsenic on telomere length, we treated NB4 cells with different concentrations of ATO. However, as long-term exposure and numerous cell doublings were required in order to observe telomere shortening, the use of high doses of arsenic was not possible because of rapid cell death. Therefore, we used low and moderate concentrations of arsenic (0.25, 0.5, and 0.75 µM). We were able to observe a dramatic erosion of telomere length in cells treated with 0.25 µM of ATO. By 19 days of treatment, the mean TRF length of NB4 cells was reduced from 2.85 to 1.96 kbp (870 bp) (A). We were also able to observe about 490-bp reduction of telomere length during 6 days of treatment at moderate concentrations of arsenic (0.5 and 0.75 µM; B).
Figure 4. Average telomere length during treatment of NB4 cells with ATO. NB4 cells were grown in presence of various concentrations of ATO (0.25, 0.5, and 0.75 µM). Genomic DNA (1.5 µg) was digested with Rsa1 and Hinf1 restriction enzymes and separated on a 0.8% agarose gel. Telomere length was determined by hybridization with a digoxigenin-labeled telomeric probe (TTAGGG)3 and chemiluminescent detection system. The peak value of TRF lengths were calculated and recorded as TLs. Numbers above each lane represent the day of treatment for each sample. L and H indicate DNA from the low and high telomere length samples, respectively. M represent fluorescent-labeled size markers.
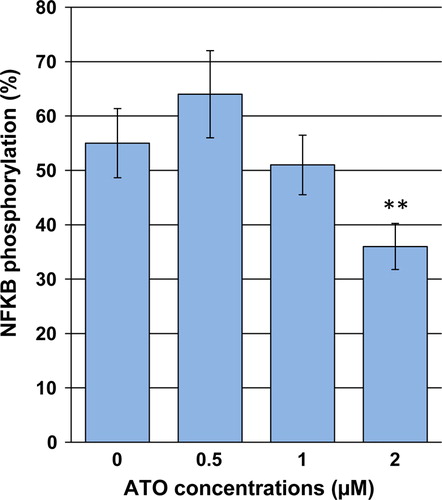
Effect of ATO on NF-κB phosphorylation
To investigate whether ATO exposure could dampen activation of NF-κB in NB4 cells, a cell-based ELISA assay was applied to evaluate the NF-κB protein phosphorylation. As shown in , treatment with ATO at 1 and 2 µM for 48 hours reduced NF-κB phosphorylation by 5.5 and 26.6%, respectively, as compared with the control.
Figure 5. Effects of ATO on activation of NF-κB in NB4 cells. Cells were exposed to desired concentrations of ATO for 48 hours. After the treatment period, the relative phosphorylated level of NF-κB was measured using a cell-based ELISA assay. Values are given as mean ± SD statistically different value of **P < 0.01 was determined compared with the control.
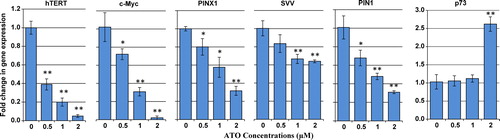
Effect of ATO on transcriptional levels of Pin1, survivin, c-Myc, hTERT, PinX1, and p73
To monitor whether ATO could modulate the transcriptional levels of Pin1, survivin, c-Myc, hTERT, PinX1, and induce the p73 message, we appraised the transcriptional levels of these genes following ATO exposure. As depicted in , the mRNA levels of Pin1, survivin, c-Myc, hTERT, and PinX1 displayed concentration-dependent reduction after 48 hours of ATO treatment. Furthermore, a significant increase in the mRNA level of p73 in response to 2 µM of ATO was observed in ATO-treated NB4 cells.
Figure 6. Effect of ATO on transcriptional levels of Pin1, hTERT, c-Myc, PinX1, survivin, and p73. The relative mRNA expression of each gene was measured using real-time reverse transcriptase-PCR in ATO-treated NB4 cells after normalizing the cycle thresholds of each triplicate against their corresponding HPRT. Values are given as mean ± SD statistically different values of *P < 0.05 and **P < 0.01 were determined compared with the control.
Discussion
Clinical trial of ATO, as a single agent, has drawn broad attention in the past decade because of its dramatic therapeutic effect on APL. ATO has proven equally effective in inducing complete clinical and molecular remission in patients with newly diagnosed and relapsed cases of APL.Citation3,Citation17 Extensive studies indicate that ATO has pleiotropic effects that make it proapoptotic, antiproliferative, and antiangiogenic.Citation18 However, the molecular mechanisms by which ATO exerts its anti-leukemic effect and induces apoptosis have not been clearly understood. So far, no single mechanism has been able to explain all of the effects seen with ATO; it probably acts at multiple levels, and with various modes of action. It has been demonstrated that ATO exerts its anti-leukemic effect both PML/RARα-dependent and PML/RARα-independent manner in APL cells. In PML/RARα-dependent, ATO cures many patients with PML/RARA-driven leukemia by targeting its PML moiety and initiating PML/RARα oncoprotein degradation, through sumoylation and ubiquitination of its moiety, which lead to its proteasome degradation.Citation19 Cell differentiation most likely occurs through the degradation of the PML-RARα product and the consequent release of the maturational block.Citation19–Citation21 In PML and PML/RARα-independent, arsenic acts on cells through a variety of mechanisms, influencing numerous intracellular signal transduction pathways and resulting in a vast range of cellular effects that include growth inhibition and apoptosis induction.Citation18,Citation22 Thus, ATO action is not limited to APL and it is used in other malignancies as well.Citation5–Citation8 It has been indicated that ATO might suppress growth and proliferation of tumor cells through inhibition of telomerase and shortening of telomere length;Citation9 however, the inhibitory mechanisms of ATO on human telomerase in tumor cells are less well understood. In this study, we mainly focused on the telomerase-dependent ATO-induced cell death, telomerase inhibition, telomere shortening, and novel postulated mechanisms underlying this process in APL cells.
As shown by the results of this study, ATO appears to be a potent telomerase inhibitor that suppresses transcriptional activation of hTERT in a dose- and time-dependent manner. Inhibition of hTERT expression and TA by therapeutic dose of ATO (2 µM) starts as early as 6 hours and by 48 hours shows more than 90% inhibition. This study shows that the treated cells continued to divide until the telomeres became critically short, and then growth arrest and apoptosis occurred in treated cells. As NB4 cells have short telomeres (the mean TRF of 2.60 kbp), a significant portion of these telomeres is expected to be at critical stage of shortening; thus, even small erosion in the average telomere length probably would be enough to trigger crisis and cell death. Respecting the progressive shortening of telomeres in ATO-exposed cells in this study (about 70–130 bp per population doubling depending on the low and moderate ATO concentrations used, see ), one might deduce that ATO might hinder proliferation and exert cell death in NB4 cells through propelling telomere shortening due to hampering telomerase enzymatic activity. In harmony, an amassing body of evidence also indicates that ATO might suppress cell growth and exerts apoptosis in tumor cells through inhibition of hTERT expression, TA and shortening of telomere length.Citation9,Citation23 Collectively, our data suggest that one of the most important key mediators of sensitivity to ATO-induced apoptosis include inhibition of TA.
A growing body of evidence implicates the striking overexpression of survivin in human malignancies of varied origin including leukemia.Citation24 This differential overexpression of survivin in tumor cells versus their normal counterparts has been thought to be associated with resistance to anti-neoplastic remediesCitation16 and decreased cell death responses.Citation25 It is interesting to note that it has been documented that survivin contributes to unconstrained tumor cell proliferation through induction of TA via stimulation of specificity protein 1 (Sp1) and c-Myc-mediated transcriptional enhancement of hTERT.Citation26 In line with this, it has been manifested that antisense ablation of survivinCitation27 or siRNA-mediated depletion of survivin expressionCitation28 causes a significant augmentation in apoptotic index. In consistence, our data delineate that ATO might impede cell growth and proliferation as well as exert programed cell death in NB4 cells through transcriptional repression of survivin. Furthermore, these outcomes imply that ATO may trammel TA and hTERT expression through suppression of survivin-induced activation of hTERT message. We next sought whether the suppressive effects of ATO on hTERT message is associated with transcriptional quelling of c-Myc. These issues indicate that ATO is capable of dampening the c-Myc mRNA.
Table 1. Nucleotide sequence of the primers used for real-time RT-PCR
In contrast, p73 has been suggested to be a negative regulator of TA via restriction of hTERT mRNA levels.Citation29 To examine the involvement of p73 in ATO-induced suppression of TA, the transcriptional level of p73 was analyzed. The results of this study indicate that ATO might trammel hTERT transcriptional levels and TA owing to stimulation of p73 expression.
There is compelling evidence that NF-κB plays pivotal roles in all stages of multilayered tumorigenesis.Citation10 Furthermore, a determinant role for NF-κB in propelling cell survival and evasion from apoptosis through induction of anti-apoptotic genes including survivin and telomerase seems plausible.Citation10 In this regard, NF-κB has attracted growing attention as an exploitable target in tumor cells and therapeutically trammeling NF-κB signaling module might open new treatment windows in order to block cell growth and proliferation.Citation10 In this setting, we next questioned whether ATO is capable to suppress the mRNA levels of survivin and telomerase through quelling the NF-κB cascade. As expressed by our issues, the inhibitory effects of ATO on transcriptional levels of survivin and telomerase in NB4 cells may be through preempting the phosphorylation and activation of the p65 subunit of the NF-κB module. Regarding the inhibitory effects of ATO on phosphorylation and activation of p65 subunit of the NF-κB cascade in our study, we next evaluated whether ATO treatment could repress the transcriptional levels of Pin1. Our data might indicate a novel inhibitory mechanism of ATO on the NF-κB signaling pathway through suppression of the Pin1-mediated activation of the p65 subunit of the NF-κB.
It has been reported that depletion of PinX1, an evolutionarily conserved nucleolar protein, by short hairpin RNA culminates in growth restriction, sensitivity to current chemotherapeutic medications as well as enfeebled tumorigenicity of telomerase-positive cells in null mice through impairing telomerase-dependent telomere length maintenance.Citation30 In this regard, PinX1-induced telomere length maintenance seems a new therapeutic target in order to disable telomerase-dependent oncogenesisCitation30 and we next evaluated whether ATO could impede the mRNA expression of PinX1. As depicted by our results, ATO exposure resulted in transcriptional repression of PinX1.
On aggregate, the results of this study indicate that ATO treatment might suppress growth and proliferation of NB4 cells through prevention of Pin1-dependent NF-κB-mediated stimulation of hTERT and thereby, continued shortening of telomeres' length which leads to programed cell death in NB4 cells. Moreover, ATO treatment might quell hTERT mRNA via stimulation of p73-induced inhibition of hTERT. In addition, the transcriptional levels of hTERT might be reduced due to suppression of survivin-dependent c-Myc-mediated transcriptional stimulation of hTERT.
Related Research Data
References
- Lo Coco F, Diverio D, Falini B, Biondi A, Nervi C, Pelicci PG. Genetic diagnosis and molecular monitoring in the management of acute promyelocytic leukemia. Blood. 1999;94(1):12–22
- Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Farretta M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295(5557):1079–82.
- Ghavamzadeh A, Alimoghaddam K, Ghaffari SH, Rostami S, Jahani M, Hosseini R, et al. Treatment of acute promyelocytic leukemia with arsenic trioxide without ATRA and/or chemotherapy. Ann Oncol. 2006;17(1):131–4.
- Jing Y, Dai J, Chalmers-Redman RM, Tatton WG, Waxman S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood. 1999;94(6):2102–11.
- Li YM, Broome JD. Arsenic targets tubulins to induce apoptosis in myeloid leukemia cells. Cancer Res. 1999;59(4):776–80.
- Rousselot P, Labaume S, Maroleau JP, Larghero J, Noguera MH, Brouet JC, et al. Arsenic trioxide and melarsoprol induce apoptosis in plasma cell lines and in plasma cells from myeloma patients. Cancer Res. 1999;59(5):1041–8.
- Akao Y, Nakagawa Y, Akiyama K. Arsenic trioxide induces apoptosis in neuroblastoma cell lines through the activation of caspase 3 in vitro. FEBS Lett. 1999;455(1–2):59–62.
- Huang XJ, Wiernik PH, Klein RS, Gallagher RE. Arsenic trioxide induces apoptosis of myeloid leukemia cells by activation of caspases. Med Oncol. 1999;16(1):58–64.
- Zhang X, Multani AS, Zhou JH, Shay JW, McConkey D, Dong L, et al. Adenoviral-mediated retinoblastoma 94 produces rapid telomere erosion, chromosomal crisis, and caspase-dependent apoptosis in bladder cancer and immortalized human urothelial cells but not in normal urothelial cells. Cancer Res. 2003;63(4):760–5.
- Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8(1):33–40.
- Altieri DC. The case for survivin as a regulator of microtubule dynamics and cell-death decisions. Curr Opin Cell Biol. 2006;18(6):609–15.
- Tran J, Master Z, Yu JL, Rak J, Dumont DJ, Kerbel RS. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc Natl Acad Sci USA. 2002;99(7):4349–54.
- Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8(1):61–70.
- Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12(6):1413–26.
- Atkinson GP, Nozell SE, Harrison DK, Stonecypher MS, Chen D, Benveniste EN. The prolyl isomerase Pin1 regulates the NF-kappaB signaling pathway and interleukin-8 expression in glioblastoma. Oncogene. 2009;28(42):3735–45.
- Hausladen DA, Wheeler MA, Altieri DC, Colberg JW, Weiss RM. Effect of intravesical treatment of transitional cell carcinoma with bacillus Calmette-Guerin and mitomycin C on urinary survivin levels and outcome. J Urol. 2003;170(1):230–4.
- Ghaffari SH, Rostami S, Bashash D, Alimoghaddam K, Ghavamzadeh A. Real-time PCR analysis of PML-RAR alpha in newly diagnosed acute promyelocytic leukaemia patients treated with arsenic trioxide as a front-line therapy. Ann Oncol. 2006;7(10):1553–9.
- Miller WH, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62(14):3893–903.
- de The H, Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer. 2010;10(11):775–83.
- Jeanne M, Lallemand-Breitenbach V, Ferhi O, Koken M, Le Bras M, Duffort S, et al. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010;18(1):88–98.
- Lallemand-Breitenbach V, Jeanne M, Benhenda S, Nasr R, Lei M, Peres L, et al. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat Cell Biol. 2008;10(5):547–55.
- Rojewski MT, Korper S, Schrezenmeier H. Arsenic trioxide therapy in acute promyelocytic leukemia and beyond: from bench to bedside. Leuk Lymphoma. 2004;45(12):2387–401.
- Zhou C, Boggess JF, Bae-Jump V, Gehrig PA. Induction of apoptosis and inhibition of telomerase activity by arsenic trioxide (As2O3) in endometrial carcinoma cells. Gynecol Oncol. 2007;105(1):218–22.
- Kamihira S, Yamada Y, Hirakata Y, Tomonaga M, Sugahara K, Hayashi T, et al. Aberrant expression of caspase cascade regulatory genes in adult T-cell leukaemia: survivin is an important determinant for prognosis. Br J Haematol. 2001;114(1):63–9.
- Tanaka K, Iwamoto S, Gon G, Nohara T, Iwamoto M, Tanigawa N. Expression of survivin and its relationship to loss of apoptosis in breast carcinomas. Clin Cancer Res. 2000;6(1):127–34.
- Endoh T, Tsuji N, Asanuma K, Yagihashi A, Watanabe N. Survivin enhances telomerase activity via up-regulation of specificity protein 1- and c-Myc-mediated human telomerase reverse transcriptase gene transcription. Exp Cell Res. 2005;305(2):300–11.
- Chen J, Wu W, Tahir SK, Kroeger PE, Rosenberg SH, Cowsert LM, et al. Down-regulation of survivin by antisense oligonucleotides increases apoptosis, inhibits cytokinesis and anchorage-independent growth. Neoplasia. 2000;2(3):235–41.
- Kami K, Doi R, Koizumi M, Toyoda E, Mori T, Ito D, et al. Downregulation of survivin by siRNA diminishes radioresistance of pancreatic cancer cells. Surgery. 2005;138(2):299–305.
- Beitzinger M, Oswald C, Beinoraviciute-Kellner R, Stiewe T. Regulation of telomerase activity by the p53 family member p73. Oncogene. 2006;25(6):813–26.
- Zhang B, Bai YX, Ma HH, Feng F, Jin R, Wang ZL, et al. Silencing PinX1 compromises telomere length maintenance as well as tumorigenicity in telomerase-positive human cancer cells. Cancer Res. 2009;69(1):75–83.