Abstract
Background
Several studies indicate that ex vivo cytokine-based expansion of cord blood (CB) CD34+ cells can induce cell cycle abnormality in expanded cells. Cycline-dependent kinase inhibitors, p21 and p57, are critical regulators of cell cycle. We investigated mRNA expression and promoter DNA methylation status of p21 and p57 genes in ex vivo expanded CD34+ cells in different culture conditions.
Materials and methods
Human CB CD34+ cells were cultured either in cytokine-supplemented liquid culture or in co-culture with mesenchymal stromal cells (co-culture system) in presence and absence of cytokine. After 14 days, expansion fold in total nucleated cells (TNCs), CD34+ cells, CD34+/CD38− cells and colony-forming units in culture (CFU-C) count was determined. Moreover, mRNA expression level and promoter DNA methylation status of p21 and p57 genes in expanded CD34+ cells were analyzed by real-time reverse transcriptase-polymerase chain reaction (PCR) and methylation-specific PCR technique, respectively.
Results
Maximum expansion of TNC, CD34+ cells, CD34+/CD38− cells and CFU-C was seen at day 10 in all three culture conditions. p21 and p57 mRNA expression level decreased in both cytokine liquid culture and co-culture system with cytokine, while in the co-culture system without cytokine, the mRNA expression of p21 and p57 genes relatively increased. In addition, promoter DNA methylation status of p21 and p57 genes did not change during expansion in all three culture conditions.
Conclusions
Co-culture of CB CD34+ cells with mesenchymal stromal cells, especially in the absence of cytokines, maintained mRNA expression of p21 and p57 genes in ex vivo expanded CD34+ cells. Moreover, transcriptional repression of p21 and p57 genes in ex vivo expanded CD34+ cells was not mediated by promoter DNA methylation.
Introduction
Human umbilical cord blood (CB) has emerged as a promising source of hematopoietic stem cells (HSC) for patients requiring allogeneic transplantation.Citation1
Ex vivo expansion of human CB HSCs using different combination of early acting cytokines is a well-known strategy to overcome stem cell dose limitation in a single CB unit.Citation2
However, several studies have indicated that ex vivo expansion of CB CD34+ cells in cytokine liquid culture has failed to improve transplantation success rates,Citation3–Citation5 and this failure has been attributed to loss of HSC function due to entrance into cell cycle.Citation6–Citation9
On the other hand, some studies indicated that mesenchymal stromal cells (MSCs) as an important component of the bone marrow microenvironment provide a ‘niche-like’ milieu for HSCs during ex vivo expansion, thereby controlling the proliferation, quiescence, and cycling of HSCs.Citation10 Consequently, co-culture of CB HSCs with a MSC feeder layer may be an ideal method to prevent cell cycle deregulation of HSCs.Citation11
The most primitive human CD34+ hematopoietic cells are quiescent, and cytokine stimulation during ex vivo culture leads to their entrance into cell cycle.Citation6 There is strong evidence that a relationship exists between cell cycle status and the engraftment ability of HSCs, since the entrance of HSCs into S/G2/M phase of cell cycle results in the loss of engraftment potential.Citation7–Citation9
The ability of HSCs to stay in the quiescent state seems to be pivotal for maintenance of their ‘stemness’ and engraftment potential.Citation12 The Cip/Kip family of cyclin-dependent kinase inhibitors (CKDIs), including p21cip1 (p21) and p57kip2 (p57) genes, plays critical roles in regulation of cell cycle kinetics and restriction of HSCs progression from G1 to S phase of the cell cycle.Citation13 Numerous studies have indicated the importance of CKDIs, p21 and p57, in controlling quiescence, proliferation and self-renewal potential of HSCs.Citation14,Citation15 Therefore, transcriptional silencing of p21 and p57 genes may result in increased cycling and exhaustion of HSCs pool.
On the other hand, methylation of the 5′ promoter region of DNA, which leads to transcriptional silencing, is one of the mechanisms involved in suppressing the expression of genes.Citation16 So, the cell cycle abnormalities in ex vivo expanded CD34+ cells may be mediated by transcriptional silencing of p21 and p57 genes via promoter DNA methylation during ex vivo expansion of CB CD34+ cells. Additionally, several studies have indicated that aberrant DNA methylation is involved in transcriptional silencing of p21 and p57 genes in various hematological malignancies, and plays a crucial role in their pathogenesis.Citation17,Citation18
Because of the critical roles of p21 and p57 genes in regulating cell cycle and in maintenance of quiescence and self-renewal capacity of HSCs, we aimed to evaluate mRNA expression and promoter DNA methylation status of these two genes in expanded CD34+ cells either in cytokine (SCF, TPO, FLT3L)-supplemented liquid culture, or in co-culture with MSCs feeder layer in the presence and absence of cytokine, and compared the results with each other.
Materials and methods
Isolation of cord blood CD34+ cells
CB samples were taken after written consent using guidelines approved by the ethics committee on the use of human subjects in the Tehran University of Medical Sciences. CB mononuclear cells (MNCs) were isolated by density gradient centrifugation on Ficoll-Hypaque (Biosciences, Sweden). The CD34+ cells were twice enriched using immunomagnetic separation with Mini MACS kit (Miltenyi Biotec, Germany). The purity of enriched CD34+ cells as determined by flow cytometery was 90% ± 6%. The viability of CD34+ cells as assessed by trypan blue dye exclusion method was consistently above 85%.
Isolation of mesenchymal stromal cells and cultivation of feeder layer
Bone marrow aspirates were taken from healthy donors after written consent. In brief, MNCs were collected from 10 ml bone marrow aspirate by density gradient centrifugation on Ficoll-Hypaque, then they were cultured in DMEM-LG supplemented with 10% fetal bovine serum (FBS) in a 75-cm2 culture flask.
After 2–3 days, non-adherent (hematopoietic) cells were removed, and the adherent cells were cultured until they reached 80% confluence. The adherent cells were harvested by 0.25% trypsin-EDTA solution, and were characterized as MSCs by osteogenic differentiation potential using a commercial kit (Chemicon, Billerica, MA, USA) and also by morphological features. These cells were further characterized by flow-cytometeric analysis, and were positive for CDw90, CD105, CD166, and CD73 markers, whereas they were negative for CD34 and CD45 markers. MSCs of passage 3 to 6 were cultured in six-well culture plates (Nunc, Denmark) at a density of 2 × 104 cells/well with DMEM-LG containing 10% FBS. After reaching 80% confluence, MSC feeder layer was used for co-culture experiments.
Ex vivo expansion of enriched cord blood CD34+ cells
Enriched CB CD34+ cells (3 × 104/ml) were cultured in six-well culture plates for 14 days in stem span serum free-medium (Stem Cell Technologies, Denmark) at 37°C under 5% CO2 humidified air in three conditions including:
(A) Cytokine liquid culture supplemented with SCF (100 ng/ml), TPO (100 ng/ml) and FLT3L (50 ng/ml) (Stem Cell Technology, Denmark).
(B) Co-culture with MSCs feeder layer (co-culture system) and the above-mentioned cytokines.
(C) Co-culture with MSC feeder layer (co-culture system) without any cytokine.
Cultures were half-fed every 3 to 4 days by harvesting half of the culture volume, and the resulting cells were used for various analyses. Collected cells were evaluated for number, viability, immunophenotyping, and clonogenic assays. In addition, CD34+ cells were re-isolated from harvested cells by Mini MACS system and were used for DNA and RNA extraction. In culture condition C, various analyses were only performed on harvested cells at day 10 of culture.
Colony-forming cell assay
To investigate the multipotency of expanded cells, 1–2 × 103 fresh CD34+ cells and 1–2 × 104 of day 10 expanded cells were cultured in 1.1 ml of complete methylcellulose medium containing recombinant cytokines (MethoCult GF+ H4434; Stem Cell Technology, Vancouver, BC, Canada) for 14 days at 37°C in a humidified air, according to manufacturer's instruction. Total number of colony-forming units in culture (CFU-C) was subsequently counted with an inverted microscope. Experiments were performed in triplicate, and clusters consisting of 40 or more cells were described as colonies.
Proliferative and immunophenotypic analysis of expanded cells
Ex vivo expansion of CB CD34+-enriched cells was evaluated by counting viable cell and by immunophenotypic analysis. Trypan Blue (Gibco BRL GmbH, Eggenstein, Germany) exclusion method was used to determine the viable hematopoietic cell number in different times in all culture conditions.
Immunophenotypic analysis for stem cell and lineage markers was conducted by flow cytometric analysis (Partec, Germany) using monoclonal antibodies against CD3, CD7, and CD19 to evaluate lymphoid lineage differentiation; CD14, CD15, and CD33 to assess myeloid differentiation; Glycophorin A to evaluate erythroid differentiation and CD34 and CD38 to evaluate stem cells markers in cells.
Flow cytometric analysis was done after incubating the harvested cells with different monoclonal antibodies conjugated to various fluorescent dyes at 4°C for 45 minutes. Isotype controls were used in each and every experiment.
Extraction of total RNA and DNA
Total RNA and DNA was extracted from fresh CB CD34+ cells (day 0) and from expanded CD34+ cells at days 3, 7, 10 and 14 of expansion in culture conditions A and B. In culture condition C, total RNA and DNA was only extracted at day 10 of culture. Total RNA extraction was performed by RNeasy Plus mini Kit (Qiagen GmbH, Hilden, Germany) or Trizol reagent (Gibco) and DNA extraction was conducted using the QIAamp DNA Mini-kit (Qiagen) according to manufacturer's protocol.
cDNA synthesis and quantitative real-time RT-PCR assay
Reverse transcription was performed on 50–500 ng of total RNA using QuantiTect Rev. Transcription Kit (Qiagen, USA) in accordance with the manufacturer's protocol.
Quantitative real-time RT-PCR was performed using the Rotor-Gene 3000 instrument (Corbett, Germany). The reaction mixture consisted of 10 µl QuantiFast SYBR Green PCR Master Mix (Qiagen), 1 µl of each primer (10 µM), 2 µl cDNA and 6 µl PCR grade water. Thermal cycler conditions were as follows: initial denaturation at 95°C for 5 minutes, 40–45 subsequent cycles of denaturation at 95°C for 15 seconds and annealing/extension at 60°C for 30 seconds. All samples were run in duplicate, and β2M was amplified as internal control to normalize target gene expression. Fold changes in gene expression among different samples were determined by 2−ΔΔCT method.
Amplification specificity was assessed by melt curve analysis. The sequence of real-time RT-PCR primers is shown in .
Table 1. Sequence of primers used for real-time RT-PCR and MSP analysis
Bisulfite modification of DNA and methylation-specific PCR
Approximately 1 µg of isolated DNA was modified by sodium bisulfite using EpiTect Bisulfite Kit (Qiagen, USA). Bisulfite converted DNA was suspended in 50 µl of TE buffer and stored at −20°C until methylation-specific PCR (MSP) analysis.
MSP analysis: Primers specific for methylated (M primer) and unmethylated (U primer) states of p21 and p57 promoter region, were designed using the online Methprimer software (http://www.urogene.org/methprimer/index1.html).
The sequence of primers is shown in . The PCR mixture (25 µl) consisted of 12.5 µl 2× Master Mix (EpiTect Msp Kit; Qiagen), 1 µl of each primer (10 µM), 30 ng bisulfite converted DNA and an appropriate volume of PCR grade water. Bisulfite converted methylated and unmethylated EpiTect PCR DNA (Qiagen, USA) were used as controls. The reactions started at 95°C for 10 minutes followed by 40 subsequent cycles at 95°C for 30 seconds, 55°C (p21, M primer), 58°C (p21, U primer; p57, M primer), 56°C (p57, U primer) for 30 seconds, 72°C for 30 seconds and a final step at 72°C for 7 minutes. PCR products were electrophoresed on 2% agarose gel and were stained with ethidium bromide.
Statistical methods
Results were reported as mean ± SD or mean ± SEM. T-test was used to evaluate statistical significance between means. A P value of <0.05 was considered statistically significant.
Results
Flow cytometric analysis of the cord blood expanded cells in three culture conditions
We examined the expression of the stem/progenitor cell markers, CD34 and CD38, on expanded cells by flow cytometry. At day 0 (before expansion), the purity of enriched CD34+ population after Mini MACS sorting was 90% ± 6%, of which 55% ± 12% expressed CD38 marker (n = 5) (A). B–1D show the percentage of CD34 and CD38 markers in day 10 expanded cells in cytokine liquid culture, in co-culture system with cytokine and in co-culture system without any cytokine, respectively.
Figure 1. Flow cytometric analysis of fresh CB CD34+ enriched cells (A) and ex vivo expanded cells in cytokine liquid culture (B), co-culture system with cytokine (C) and co-culture system without any cytokine (D). Specific staining was performed with anti-CD34-FITC and anti-CD38-PE antibodies. (A) CD34: 94.25%; CD38: 54.63%. (B) CD34: 71.63%; CD38: 67.38%. (C) CD34: 79.48%; CD38: 65.07%. (D) CD34: 77.22%; CD38: 64.42%.
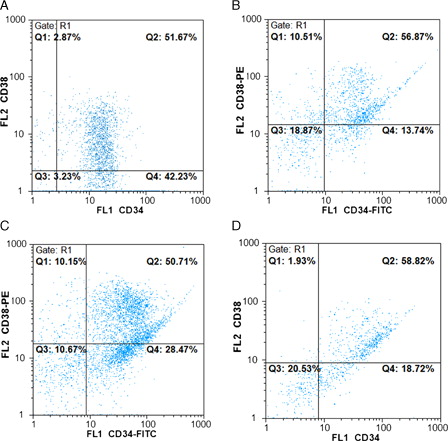
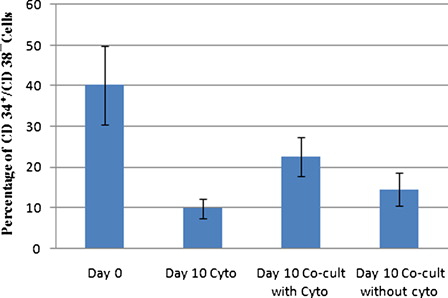
As shown in , the mean percentage of CD34+/CD38− cells in fresh CB CD34+ isolated cells (day 0) was 40.2 ± 9.7%, whereas the mean percentage of CD34+/CD38− cells at day 10 expanded cells decreased in all three culture conditions and reached 9.8 ± 2.3, 22.6 ± 4.8, and 14.5 ± 4.1% in cytokine liquid culture, in co-culture system with cytokine, and in co-culture system without cytokine, respectively. It is obvious that co-culture system with cytokine preserved a higher percentage of CD34+/CD38− hematopoietic cells than cytokine liquid culture (P < 0.01). After 10 days of expansion, relative differentiation was seen and lineage marker expression on expanded cells in cytokine liquid culture was higher than co-culture system with cytokine.
Figure 2. Mean percentage of CD34+/CD38− cells among fresh CB CD34+ isolated cells and expanded cells at day 10 in three culture conditions (n = 5).
Evaluation of ex vivo expansion of cord blood CD34+-enriched cells in cytokine liquid culture and in co-culture system with and without cytokines
We evaluated ex vivo expansion of CB CD34+-enriched cells in all culture conditions. A and 3B show the fold increase in number of total nucleated cells (TNCs) and CD34+ cells, respectively, during 14 days of ex vivo expansion in cytokine liquid culture, in co-culture system with cytokine and in co-culture system without cytokine (n = 5). Maximum expansion was observed at day 10 of culture in all culture conditions. In cytokine supplemented liquid culture, the mean fold change in TNC and CD34+ cell number was 46.23 ± 17.84 and 26.76 ± 8.39, respectively. Moreover, the mean fold change in TNC and CD34+ cell number was 71.92 ± 13.05 and 64.32 ± 10.9, respectively, in co-culture system with cytokine. On the other hand, in co-culture system without cytokine, TNC and CD34+ cell number was increased up to four folds.
Figure 3. Mean fold change of TNC (A) and CD34+ cells (B) in cytokine liquid culture and in co-culture system with and without cytokine.
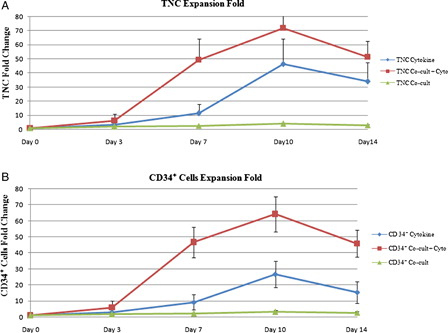
The expansion rate of TNC and CD34+ cells in co-culture system with cytokine was significantly higher compared with either cytokine liquid culture ((P < 0.05) for TNC and (P < 0.001) for CD34+ cells) or co-culture system without cytokine ((P < 0.0001) for TNC and CD34+ cells).
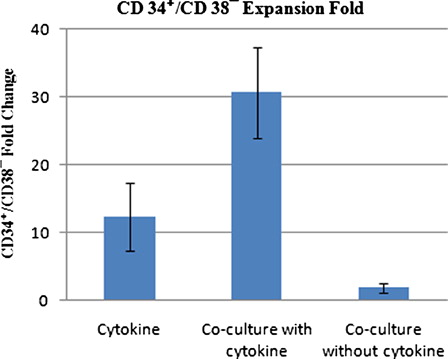
We also determined the expansion of CD34+/CD38− primitive hematopoietic cells after 10 days of ex vivo expansion. As shown in , at day 10, the CD34+/CD38− cells increased 12.3 ± 5.1-fold in cytokine liquid culture, 30.65 ± 6.7-fold in co-culture system with cytokine and 1.8 ± 0.7-fold in co-culture system without cytokine, as compared with day 0 (n = 5). Therefore, the co-culture system with cytokine resulted in higher fold increase of CD34+/CD38− cells compared with either cytokine liquid culture (P < 0.001) or co-culture system without cytokine (P < 0.00001).
Figure 4. Mean fold increase of CD34+/CD38− cells in cytokine supplemented liquid culture and in co-culture system with and without cytokine on day 10 of expansion in three culture conditions.
Evaluation of clonogenic potential
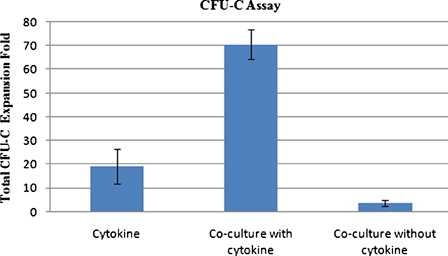
Clonogenic potential of the ex vivo expanded cells was evaluated by CFU assay. The total fold increase in CFU-C number at day 10 of culture was 19.3 ± 7.3, 70.5 ± 6.24, and 3.5 ± 1.3 for cytokine liquid culture, co-culture system with cytokine, and co-culture system without cytokine (n = 4), respectively (). The presence of both MSC and cytokine in culture resulted in higher clonogenic potential of expanded cells after 10 days of expansion. Therefore, the co-culture system with cytokine produced significantly higher fold increase in CFU-C count than co-culture system without cytokine (P < 0.0001) and cytokine liquid culture (P < 0.001).
Figure 5. CFU assay of ex vivo expanded hematopoietic cells at day 10 of culture.
mRNA expression of p21 and p57 genes during ex vivo expansion of CB CD34+ cells
Real-time RT-PCR results showed statistically significant differences in mRNA expression of p21and p57 genes between fresh CD34+ cells and expanded CD34+ cells in different days of expansion in cytokine liquid culture. As shown in A and 6B, the mRNA expression of p21 and p57 genes in expanded CD34+ cells gradually decreased with culture and reached the lowest expression at day 10 of expansion relative to day 0 (fold change p21 = 0.07 ± 0.014, P < 0.001; fold change p57 = 0.04 ± 0.006, P < 0.001 (n = 5)); however, the mRNA expression of p21 but not p57 increased relatively on day 14. A relatively similar pattern of p21 and p57 mRNA expression was seen in co-culture system with cytokine, so that the lowest mRNA expression of p21 and p57 genes was seen at day 7 and day 10 of expansion, respectively, relative to day 0 (fold change p21 = 0.21 ± 0.014, P < 0.001, (n = 5)) and (fold change p57 = 0.13 ± 0.008, P < 0.001 (n = 5)); nevertheless, the expression of p21 increased relatively on day 14.
Figure 6. mRNA expression fold changes of p21 (A) and p57 (B) in expanded CD34+ cells relative to fresh CB CD34+ cells in cytokine liquid culture and in co-culture system with cytokine. Data are plotted as mean ± SD of 2−ΔΔCT, which is directly proportional to the relative gene expression.
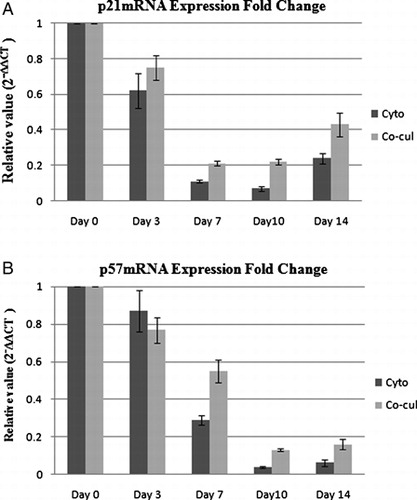
As stated, the mRNA expression of p21 and p57 genes decreased in both cytokine liquid culture and co-culture system with cytokine; however, statistical analysis showed that the relative mRNA expression of p21 and p57 was significantly higher in co-culture system with cytokine than cytokine liquid culture (P < 0.01, for p21 and P < 0.001 for p57).
However, in co-culture system without cytokine, the mRNA expression of p21 and p57 genes in expanded cells slightly increased relative to fresh CD34+ cells, but the difference in expression level was only significant for p21 (fold change p21 = 1.71 ± 0.12, P < 0.01 and fold change p57 = 1.13 ± 0.08, P > 0.05, (n = 5)). Therefore, the co-culture system without cytokine augmented or at least retained the expression of CDKIs, p21 and p57, in expanded cells.
Methylation analysis of p21 and p57 genes' promoter during ex vivo expansion of CB CD34+ cell
We investigated promoter DNA methylation status of p21 and p57 genes in freshly isolated CD34+ cells and expanded CD34+ cells using MSP technique.
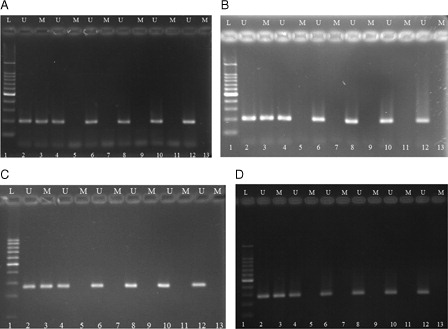
Our results showed that p21 and p57 promoter region was unmethylated in fresh CB CD34+ cells and remained unmethylated in expanded CD34+ cells at different days of expansion in cytokine liquid culture and in co-culture system with cytokine (n = 5) (). Similarly, promoter of p21 and p57 genes remained unmethylated in expanded cells at co-culture system without cytokine. Therefore, our results demonstrated that transcriptional repression of p21 and p57 genes in ex vivo expanded CD34+ cells was not mediated by promoter DNA methylation.
Figure 7. Methylation-specific PCR of bisulfite-modified DNA using primer pairs specific for methylated and unmethylated p21 and p57 genes' promoter sequence. (A, B) representing p21 MSP results and (C, D) representing p57 MSP results for expanded cells in cytokine liquid culture and in co-culture system with cytokine, respectively. M, primers specific for methylated DNA; U, primers specific for unmethylated DNA. 1, DNA ladder; 2, unmethylated DNA control; 3, methylated DNA control; 4, 6, 8, 10, and 12 illustrate DNA amplification at days 0, 3, 7, 10, and 14 of culture, respectively, with unmethylated primers; 5, 7, 9, 11, and 13 had no band indicating lack of DNA amplification for expanded cells at days 0, 3, 7, 10, and 14 of culture, respectively, with methylated primers.
Discussion
p21 and p57, as cell cycle inhibitors, play a crucial role in regulating cell cycle kinetics and in maintaining quiescence in the HSCs.Citation13–Citation15,Citation19 In this study, we expanded CB CD34+ cells in three different culture conditions for 14 days and compared them in terms of mRNA expression and promoter DNA methylation status of p21 and p57 genes in expanded cells along with expansion fold in TNC, CD34+ cells, CD34+/CD38− cells, and CFU-C count.
Expansion results showed that the mean fold increase of TNC, CD34+ cells, CD34+/CD38− cells, and total CFU-C count was higher in co-culture system with cytokine than cytokine liquid culture and co-culture system without cytokine. In agreement with our results, Walenda et al. and Da Silva et al. showed that MSCs and cytokines have synergistic effects in ex vivo expansion of HSCs, and their synergistic effect resulted in higher expansion folds in co-culture with MSCs.Citation11,Citation20 Also, in agreement with our results, Jang et al.Citation21 showed that in co-culture system without cytokine, CD34+ cells have limited proliferative and clonogenic potential.
Our study showed decreased mRNA expression of p21 and p57 genes in expanded CD34+ cells in cytokine supplemented liquid culture and it was thus consistent with the study of Araki et al.Citation22, Holmes et al.Citation23, and Ko et al.Citation24 that showed decreased mRNA expression of p21 and p57 genes in ex vivo expanded cells in cytokine supplemented culture. Recently, a study by Yamazaki et al.Citation6 showed that cytokine stimulation led to re-entry of HSCs into the cell cycle and also reduced expression of p57 in HSCs.
In this study, mRNA expression of p21 and p57 genes in expanded CD34+ cells also decreased in co-culture system with cytokine. However, the mRNA expression of p21 and p57 genes in expanded CD34+ cells was significantly higher in co-culture system with cytokine than cytokine liquid culture. This result may be explained by higher fold increase of primitive CD34+/CD38− cells in co-culture system with cytokine than cytokine liquid culture (30.65 ± 6.7 vs. 12.3 ± 5.1); because the study by Passegué et al.Citation25 showed that the highest mRNA expression of p21 and p57 genes is found in most primitive hematopoietic cells.
Our results was in agreement with Kadereit et al.Citation26 study that showed expansion of CB CD34+/CD38− cells on MSCs feeder layer resulted in maintenance of p21 expression in expanded hematopoietic cells.
On the other hand, in co-culture system without cytokine, the mRNA expression of p21 and p57 genes in expanded cells was higher in comparison to fresh CD34+ cells.
In agreement with our results, Moreno et al. reported that in the absence of cytokines, the levels of p16 and p21 increased at CD34+ expanded cells, and suggested that such cell cycle regulators are directly involved in the control of proliferation in hematopoietic progenitor cells.Citation27
Stier et al.Citation15 demonstrated that ex vivo silencing of p21 resulted in clonal expansion of HSCs. This is in agreement with our observations where expanded HSCs had less p21 mRNA expression with the highest proliferation capacity in cytokine supplemented cultures. On the other hand, expanded HSCs in the co-culture system without cytokine had the highest p21 mRNA expression, and their proliferation capacity was low.
The possible mechanisms by which MSCs regulate the cell cycle and quiescence of HSCs leading to increased mRNA expression of CDKIs p21 and p57 in HSCs may be related to production of growth factors such as SDF-1 (CXCL12), TGFb and angiopoietin-1 by MSCs.Citation28–Citation31
Our results showed that promoter regions of p21 and p57 genes were unmethylated in fresh CB CD34+ cells and remained unmethylated in expanded CD34+ cells in all culture conditions. Therefore, the observed transcriptional repression of p21 and p57 genes in ex vivo expanded cells in our study may be mediated by other epigenetic mechanisms such as histone deacetylation or regulatory micro RNA. Promoter DNA methylation of p57 and p21 genes is involved in pathogenesis of hematologic malignancies such as acute lymphoblastic leukemia and lymphoma.Citation17,Citation18,Citation32,Citation33 In our study, promoter DNA methylation of p21 and p57 genes was not seen in ex vivo expanded cells, indicating that our culture conditions and cytokine cocktail had no effect on methylation status of these genes. However, decreased mRNA expression of these tumor suppressor genes in ex vivo expanded cells may warrant further investigation.
Conclusion
The presence of MSCs in co-culture system, especially in the absence of cytokines, maintained mRNA expression of p21 and p57 genes in ex vivo expanded CD34+ cells. Moreover, transcriptional repression of p21 and p57 genes in ex vivo expanded CD34+ cells was not mediated by promoter DNA methylation.
Acknowledgements
The authors thank Tehran University of Medical Sciences and Iranian Blood Transfusion Organization Research Center for research because of their financial supports. Also, they are so grateful to the referees of this paper because their insightful comments caused this paper a significant improvement in quality.
References
- Stanevsky A, Goldstein G, Nagler A. Umbilical cord blood transplantation: pros, cons and beyond. Blood Rev. 2009;23(5):199–204.
- Tung S, Parmar S, Robinson S, De Lima M, Shpall E. Ex vivo expansion of umbilical cord blood for transplantation. Best Pract Res Clin Haematol. 2010;23(2):245–57.
- McNiece IK, Almeida-Porada G, Shpall EJ, Zanjani E. Ex vivo expanded cord blood cells provide rapid engraftment in fetal sheep but lack long-term engrafting potential. Exp Hematol. 2002;30(6):612–6.
- Habibian HK, Peters SO, Hsieh CC, Wuu J, Vergilis K, Grimaldi CI, et al. The fluctuating phenotype of the lymphohematopoietic stem cell with cell cycle transit. J Exp Med. 1998;188(2):393–8.
- Wulf-Goldenberg A, Eckert K, Fichtner I. Cytokine-pretreatment of CD34(+) cord blood stem cells in vitro reduces long-term cell engraftment in NOD/SCID mice. Eur J Cell Biol. 2008;87(2):69–80.
- Yamazaki S, Iwama A, Takayanagi S, Morito Y, Eto K, Ema H, et al. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 2006;25(15):3515–23.
- Gothot A, van der Loo JCM, Clapp DW, Stour EF. Cell cycle-related changes in repopulating capacity of human mobilized peripheral blood CD34+ cells in non-obese diabetic/severe combined immune-deficient mice. Blood. 1998;92(8):2641–9.
- Wilpshaar J, Falkenburg JHF, Tong X, Noort WA, Breese R, Heilman D, et al. Similar repopulating capacity of mitotically active and resting umbilical cord blood CD34+ cells in NOD/SCID mice. Blood. 2000;96(6):2100–7.
- Glimm H, Oh ILH, Eaves CJ. Human hematopoietic stem cells stimulated to proliferate in vitro lose engraftment potential during their S/G2/M transit and do not reenter G0. Blood. 2000;96(13):4185–93.
- Jing D, Fonseca AV, Alakel N, Fierro FA, Muller K, Bornhauser M, et al. Hematopoietic stem cells in co-culture with mesenchymal stromal cells-modeling the niche compartments in vitro. Haematologica. 2010;95(4):542–50.
- Walenda T, Bork S, Horn P, Wein F, Saffrich R, Diehlmann A, et al. Co-culture with mesenchymal stromal cells increases proliferation and maintenance of haematopoietic progenitor cells. J Cell Mol Med. 2010;14(1–2):337–50.
- Iriuchishima H, Takubo K, Matsuoka S, Onoyama I, Nakayama KI, Nojima Y, et al. Ex vivo maintenance of hematopoietic stem cells by quiescence induction through Fbxw7α overexpression. Blood. 2011;117(8):2373–7.
- Pietras EM, Warr MR, Passegué E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195(5):709–20.
- Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y, et al. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9(3):262–71.
- Stier S, Cheng T, Forkert R, Lutz C, Dumbkowski DM, Zhang JL, et al. Ex vivo targeting of p21Cip1/Waf1 permits relative expansion of human hematopoietic stem cells. Blood. 2003;102(4):1260–6.
- Paszkowski J, Whitham SA. Gene silencing and DNA methylation processes. Curr Opin Plant Biol. 2001;4(2):123–9.
- Shen LL, Toyota M, Kondo Y, Obata T, Daniel S, Pierce S, et al. Aberrant DNA methylation of p57KIP2 identifies a cell-cycle regulatory pathway with prognostic impact in adult acute lymphocytic leukemia. Blood. 2003;101(10):4131–6.
- Roman-Gomez J, Castillejo JA, Jimenez A, Gonzalez MG, Moreno F, Rodriguez MdelC, et al. 5′ CpG island hypermethylation is associated with transcriptional silencing of the p21CIP1/WAF1/SDI1 gene and confers poor prognosis in acute lymphoblastic leukemia. Blood. 2002;99(7):2291–6.
- Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, et al. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9(3):247–61.
- Da Silva CL, Gonçalves R, Crapnell KB, Cabral JM, Zanjani ED, Almeida-Porada G, et al. A human stromal-based serum-free culture system supports the ex vivo expansion/maintenance of bone marrow and cord blood hematopoietic stem/progenitor cells. Exp Hematol. 2005;33(7):828–35.
- Jang YK, Jung DH, Jung MH, Kim DH, Yoo KH, Sung KW, et al. Mesenchymal stem cells feeder layer from human umbilical cord blood for ex vivo expanded growth and proliferation of hematopoietic progenitor cells. Ann Hematol. 2006;85(4):212–25.
- Araki H, Yoshinaga K, Boccuni P, Zhao Y, Hoffman R, Mahmud N. Chromatin-modifying agents permit human hematopoietic stem cells to undergo multiple cell divisions while retaining their repopulating potential. Blood. 2007;109(8):3570–8.
- Holmes T, Yan F, Ko KH, Nordon R, Song E, O'Brien TA, et al. Ex vivo expansion of cord blood progenitors impairs their short-term and long-term repopulating activity associated with transcriptional dysregulation of signalling networks. Cell Prolif. 2012;45(3):266–78.
- Ko KH, Holmes T, Palladinetti P, Song E, Nordon R, O'Brien TA, et al. GSK-3beta inhibition promotes engraftment of ex vivo-expanded hematopoietic stem cells and modulates gene expression. Stem Cells. 2011;29(1):108–18.
- Passegué E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202(11):1599–611.
- Kadereit S, Deeds LS, Haynesworth SE, Koc ON, Kozik MM, Szekely E, et al. Expansion of LTC-ICs and maintenance of p21 and BCL-2 expression in cord blood CD34(+)/CD38(−) early progenitors cultured over human MSCs as a feeder layer. Stem Cells. 2002;20(6):573–82.
- Alvarado-Moreno A, Chávez-González A, Cérbulo A, Arriaga L, Mayani H. In vitro cell cycle dynamics of primitive hematopoietic cells from human umbilical cord blood. Hematology. 2010;15(1):11–20.
- Liang X, Su YP, Kong PY, Zeng DF, Chen XH, Peng XG, et al. Human bone marrow mesenchymal stem cells expressing SDF-1 promote hematopoietic stem cell function of human mobilised peripheral blood CD34+ cells in vivo and in vitro. Int J Radiat Biol. 2010;86(3):230–7.
- Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118(2):149–61.
- Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. 2008;205(4):777–83.
- Scandura JM BP, Massagué J, Nimer SD. Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc Natl Acad Sci USA. 2004;101(42):15231–6.
- Li Y, Nagai H, Ohno T, Yuge M, Hatano S, Ito E, et al. Aberrant DNA methylation ofp57 KIP2 gene in the promoter region in lymphoid malignancies of B-cell phenotype. Blood. 2002;100(7):2572–7.
- Go JH. Methylation analysis of cyclin-dependent kinase inhibitor genes in primary gastrointestinal lymphomas. Mod Pathol. 2003;16(8):752–5.