Abstract
In chronic myeloid leukemia (CML), epigenetic modifications such as promoter hypermethylation and inactive histone modification are known mechanisms of drug resistance. In our study, we investigated the roles of promoter hypermethylation of BIM and BID genes and H3K27me3 histone modification on imatinib resistance.
We detected higher expression levels of BIM and BID genes and lower expression levels of EZH2, EED2, SIRT1, and SUZ12 genes in imatinib-resistant K562/IMA-3 cells compared to imatinib-non-resistant K562 cells. While we determined the EZH2 and DNMT enzymes as bounded to the promoter of the BIM gene, we did not detect hypermethylation of this promoter. We also found the H3K27me3 histone modification promoter of BIM and BID genes in both cell lines. In conclusion, our results support the notion that DNA promoter methylation may be formed independently from EZH2-H3K27me3 and pro-apoptotic BIM and BID genes are not methyllated in the imatinib resistance of CML cells.
Introduction
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disease of hematopoetic stem cells. The bcr-abl oncoprotein which is formed by the reciprocal translocation of chromosomes 9 and 22 plays a role in disease pathogenesis.Citation1 CML shows a biphasic clinic course with chronic and accelerated phases, followed by the blastic crisis phase.Citation2,Citation3 Imatinib mesylate is a novel synthetic tyrosine kinase inhibitor successfully used for the treatment of CML patients.Citation4 It binds to the inactive form of Bcr-Abl and inhibits its transition from active to inactive form. While most patients with chronic phase of the disease can be cured with imanitib, resistance against imatinib is the most significant problem in imatinib treatment especially in patients with blastic crisis.Citation5 In our previous studies, we showed that mechanisms of imatinib resistance include changes in the structure and levels of BCR/ABL gene, inhibition of apoptotic signaling pathways and aberrant ceramide metabolism.Citation6,Citation7 Some of the epigenetic modifications are mechanisms of drug resistance formed independently from Bcr-Abl in CML. DNA methylation and histone modifications are two important epigenetic mechanisms.Citation8,Citation9 Methylation of the promoter region can also cause silencing of the genes.Citation10 Polycomb group proteins (PcG) are epigenetic regulators consisting of multimeric proteins and modified histone proteins. Polycomb repressive complex 4 (PRC4) is one of the subgroups of PcG proteins,Citation11 and is expressed especially in embryonic and cancer cells. PRC4 is comprised of embryonic ectoderm development (EED2), enhancer of zeste homolog (EZH1), EZH2, stress-sensing protein1 (SIRT1), and suppressor of zeste 12 (SUZ12) proteins.Citation12,Citation13 EZH2 is a methyltransferase which adds three methyl groups at the specific lysine residue on H3 histone proteins.Citation14 This modification is a marker of an inactive chromatin state. In cancer cells, EZH2-mediated H3K27 trimethylation can selectively cause DNA methylation and inhibit gene expression.Citation15,Citation16
This study was designed to examine the roles of epigenetical regulations in imatinib resistance by determining the expression levels of BIM and BID pro-apoptotic genes in imatinib-sensitive and -resistant cells and examining the mechanisms of epigenetic regulation through PRC4 protein complex.
Methods
Cell lines
Human K562 CML cells were obtained from German Collection of Microorganisms and Cell Cultures. K562 cells were maintained in RPMI1640 medium supplemented with 100 units/ml penicilin, 100 g/ml streptomycin (Gibco, Burlington, Canada) and, 10% foetal bovine serum (Sigma, St Louis, MO, USA) and incubated at 37°C in a humidified 5% CO2 incubator. K562/IMA-3 cells, which were able to grow in the presence of 3 µM imatinib, were generated from K562 cells by stepwise increasing concentrations of imatinib.
Genomic DNA Isolation
Genomic DNA was isolated from K562/IMA-3 and K562 cells by using QIAMP DNA mini kit (Qiagen, Hilden, Germany) as described by the manufacturer. Briefly, 1.5 × 106 cells/ml were lysed with the enzyme, and nucleic acids were stabilized. After adding alcohol, lysates were loaded on to the QIAamp spin column and wash buffers were used to remove impurities. Then DNA was eluted in water or low-salt buffer.
Reverse transcriptase polymerase chain reaction analysis
Total RNA was extracted with RNeasy Mini kit (Sigma, St Louis, MO, USA; A.B.D). cDNA was synthesized from 2 µg of RNA, using oligo (dT) primers and Revert Aid™ First Strand cDNA Synthesis Kit (Fermentas, Vilnius, Lithuania) as described by the manufacturer. Removal of DNA from RNA samples was achieved by DNase treatment (DNA-free™ kit, Ambion Inc., Ambion, CA, USA). The primers designed for EED2, SUZ12, SIRT1, EZH2, BID, and BIM are shown in . Polymerase chain reactions (PCRs) were carried out in a reaction mixture containing 10× buffer, MgCl2 (Fermentas, Vilnius, Lithuania), dNTP mixture (MBI Fermentas, Vilnius, Lithuania), and Taq polymerase (MBI Fermentas, Vilnius, Lithuania) using the cycling conditions; 3 minutes at 95°C, 30 seconds at 94°C, 30 seconds at the annealing temperature, and 3 minutes at 72°C. PCR products were separated on 1% agarose gel electrophoresis, stained with ethidium bromide and visualized under ultraviolet (UV) light.
Table 1. Primer Sequences of Genes for PCR Amplification
Real-time PCR
mRNA levels of EZH2, EED2, SUZ12, SIRT1, BIM, and BID genes were determined in K562/IMA-3 and K562 cells by real-time PCR. Expression level of GAPDH was used as the internal positive control. PCR was carried out in a reaction mixture containing 1× Lightcycler-DNA Master SYBR Green I (Roche Diagnostics, Mannheim, Germany), 0.125 µM primer oligonucleotides and 3.5 mM MgCl2 using the cycling conditions: 5 minutes at 65°C, 30 seconds at 60°C, and 30 seconds at 72°C (ABI Prism 7700 Sequence Detection System, Applied Biosystems, Foster City, CA, USA). Each reaction was performed in triplicates. Comparative Ct (ΔΔCt) method was used for the relative quantification of the target gene expression.Citation17
Methylation-specific PCR
Methylation status of the promoter regions of BIM and BID genes was evaluated by methylation-specific PCRs (MS-PCR), as described previously (12). Briefly, 1 µg of genomic DNA was modified by a reaction containing sodium bisulphide under the conditions that convert all unmethylated cytosines to urasil (CpG Genome DNA modification kit; Chemicon, Billerica, MA, USA). After that bisulphide-modified DNA was amplified with primers, specific for unmethylated (U) and methylated BIM and BID genes' promoter regions. As a positive control ‘CpGenome Universal Methylated DNA’ (Millipore, Billerica, MA, USA) was used. PCR amplification of the BIM gene (10 minutes at 94°C; 1 minutes at 94°C; 1 minutes at 56°C; 1 minutes at 72°C; and 10 minutes at 72°C) was performed with the primers given in .
Table 2. Primer Sequences of BID and BIM Genes for MS-PCR Amplification
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) experiments were performed according to the procedure of ‘The Simple ChIP Enzymatic Chromatin IP Kit (Magnetic Beads, Cell Signalling, Danvers, MA, USA). K562 and K562/IMA-3 cells (4 × 107) were treated with 1% formaldehye which serves to fix protein–DNA interactions in the cells. The cells were then lysed with cell lysis buffer on ice and chromatin was harvested and fragmented using sonication. Immunoprecipitation was performed using antibodies specific to EZH2 (Active Motif, eyalet, Carlsbad, CA, USA), DNMT1 (Active Motif), Tri-Methyl-Histone H3 (Cell Signalling) and ChIP Grade Protein G Magnetic Beads. After the reversal of protein–DNA cross-links, DNA was purified using DNA purification spin columns provided in the kit. After ChIP experiments PCR amplification of the BIM gene (5 minutes at 95°C; 30 seconds at 95°C; 30 seconds at 55°C; and 30 seconds at 72°C) was performed with the primers, sense 5′-CTGGTCTGCAGTTTGTTGGA-3′ and antisense 5′-GGTGGCTGCAAGAATCAAGT-3′. PCR amplification of the BID gene (5 minutes at 95°C; 30 seconds at 95°C; 30 seconds at 60°C; and 30 seconds at 72°C) was performed with the primers, sense 5′-GGCTTTGTTGTGTCCTCTCC-3′ and antisense 5′-TGTCTGCGGTGCTGGAAA-3′. H3 Histone was used as a positive control and amplified with primer sets which are specific to the human ribosomal protein L30 (RPL 30) gene locus while normal rabbit IgG was used as a negative control and amplified with primer sets that were specific to the mouse RPL 30 gene locus. PCR products were run on 2% agarose gel electrophoresis, stained with ethidium bromide, and visualized under UV light.
Results
Expression Levels of EZH2, SUZ12, SIRT1, EED2, BIM, and BID genes in K562 and K562/IMA-3 cells
Expression levels of EZH2, SUZ12, SIRT1, EED2, BIM, and BID genes were determined both in K562 and in K562/IMA-3 cells with conventional PCR as shown in . SIRT1 and EED2 isoform of EED genes were expressed only in PRC4 subgroup of PcG protein (12, 13). In this study, it was shown that PRC4 group proteins are expressed both in K562 and K562/IMA-3 cells (). In order to confirm conventional PCR results, expression levels of EZH2, SUZ12, SIRT1, EED2, BIM, and BID genes were examined by quantitative real-time PCR. The average 2−ΔΔCt value indicated that expression levels of EZH2, SIRT1, EED2, and SUZ12 genes in K562/IMA-3 cells were significantly decreased as compared with the parental-sensitive counterparts, and normalized to the internal positive control. Interestingly, expression levels of pro-apoptotic BIM and BID genes were increased in K562/IMA-3 cells compared to the parental-sensitive K562 cells ().
Figure 1. RT-PCR results of PcG, BID, and BIM genes in K562 and K562/IMA-3 cell lines. (A) RT-PCR results of the BIM gene: (1) M:50 bp, (2) No DNA, (3) K562 cells, (4) K562/IMA-3 cells, (5) positive control, (6) negative control. (B) RT-PCR results of the EED2 gene: (1) M:50 bp, (2–3) positive control, (4) K-562 cells, (5) K-562/IMA-3 cells, (6) negative control. (C) RT-PCR results of the BID gene: (1) M:50 bp, (2–4) positive control, (5) K562 cells, (6) K562/IMA-3 cells, (7) negative control. (D) RT-PCR results of the SUZ12 gene: (1) M:50 bp, (2) positive control, (3) negative control, (4) K562 cells, (5) K562/IMA-3 cells, (E) RT-PCR results of SIRT1 gene: (1) M:50 bp, (2) K562 cells, (3) K562/IMA-3 cells, (4) positive control, (5) negative control. (F) RT-PCR results of the EZH2 gene: (1) M:50 bp, (2–3) positive control, (4) K562 cells, (5) K562/IMA-3 cells, (6) negative control. (G) RT-PCR results of GAPDH gene: (1) M:50 bp, (2–3) positive control, (4–5) K-562 cells, (6–7) K562/IMA-3 cells, (8) negative control.
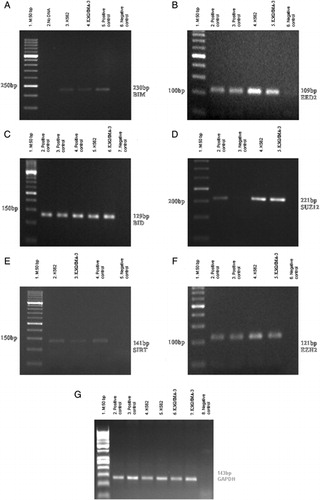
Figure 2. PcG, BID, and BIM genes expressions were determined in K562 and K562/IMA-3 cell lines by real-time RT-PCR.
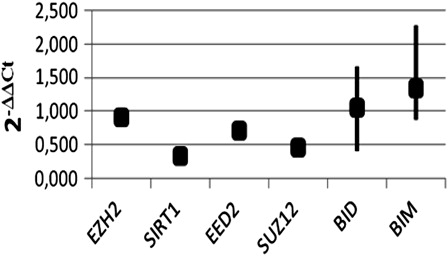
PCR results after ChIP experiments
H3K27me3 modification at BIM and BID genes promoters were determined in K562 and K562/IMA-3 cell lines by ChIP analyses ().
Figure 3. H3K27me3 histone modification on promoter region of BIM and BID genes in K562/IMA-3 cell lines was determined by ChIP-PCR. (A) (1) negative control, (2) positive control, (3) 2% input DNA, (4) M:50 bp, (5–6) K562/IMA-3 cell lines, (7) no DNA. (B) (1) M:50 bp, (2) negative control, (3) positive control, (4) 2% input DNA, (5) K562/IMA-3 cell line, (6) no DNA.
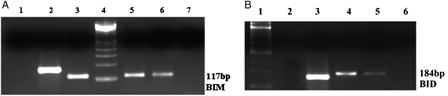
EZH2 and DNMT1 enzymes were found as bounded to the BID gene promoter region in K562 but not in imatinib-resistant K562/IMA-3 cells. Also H3K27me3 modification was detected in K562/IMA-3 cells ().
Figure 4. In K562 cell, EZH2 and DNMT1 enzymes were found as bounded to the BID gene promoter region, but in imatinib-resistant K562/IMA-3 cells these enzymes were not seen as bounded to the BID gene promoter. (1) K562 cells BID (+) control, (2) K562 cells BID (−) control, (3) K562 BID 2% input DNA, (4) M: 50 bp, (5) K562 cells EZH2 BID, (6) K562 cells Dnmt1 BID, (7) K562 cells H3K27me3 BID, (8) K562/IMA-3 cells BID (+) control, (9) K562/IMA-3 cells BID (−) control, (10) K562/IMA-3 cells BID 2% input DNA, (11) K562/IMA-3 cells EZH2 BID, (12) K562/IMA-3 cells Dnmt1 BID, (13) no DNA.
EZH2 and DNMT1 enzymes were determined as bounded to the BIM gene promoter region in both cells ().
Figure 5. EZH2 and DNMT1 enzymes were determined as bounded to the BIM gene promoter region in both cell lines. (A) (1) K562 cells BID 2% input DNA, (2) K562 cells Dnmt1 BIM, (3) K562 cells EZH2 BIM, (4) K562 cells H3K27me3 BIM, (5) K562 cells BIM (+) control, (6) K562 cells BIM (–) control, (7) M: 100 bp. (B) (1) M: 100 bp, (2) K562/IMA-3 cells BIM 2 % input DNA, (3) no DNA, (4) K562/IMA-3 cells Dnmt1 BIM, (5) K562/IMA-3 cells EZH2 BIM, (6) K562/IMA-3 cells H3K27me3 BIM, (7) K562/IMA-3 cells BIM (+) control, (8) K562/IMA-3 BIM (–) control.
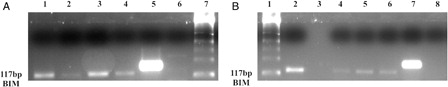
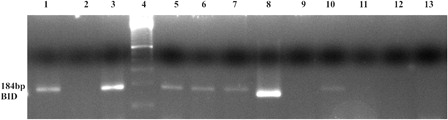
BIM and BID gene promoter regions are unmethyllated in K562 and K562/IMA-3 cells
Methylation status of the promoter regions of BIM and BID genes was evaluated by MS-PCRs. Our results revealed that BIM and BID gene promoter regions were unmethyllated homozygously in both cells despite finding DNMT1 bounded to these two proapoptotic gene promoters ().
Figure 6. BIM and BID gene promoter regions are unmethylated in K562 and K562/IMA-3 cell lines. (A) (1) M:50 bp, (2) K562 cells BIM unmethylated band, (3) K562 cells BIM methylated sample, (4) K562/IMA-3cells BIM unmethylated band, (5) K562/IMA-3cells BIM methylated sample, (6) peripheral blood sample BIM unmethylated band, (7) peripheral blood sample BIM methylated sample, (8) (+) control BIM unmethylated band, (9) (+) control BIM methylated band. (B) (1) M:50 bp, (2) unmethylated negative control, (3) K562 cells BID unmethylated band, (4) K562 /IMA-3 cells BID unmethylated band, (5) unmethylated positive control, (6), K562 cells BID methylated sample, (7) K562 /IMA-3 cells BID methylated sample, (8) methylated positive control.

Discussion
The aim of this study was to investigate epigenetic differences of BIM and BID pro-apoptotic genes between imatinib-resistant and non-resistant CML cells. For this reason, we determined methylation status and H3K27me3 modification of BIM and BID pro-apoptotic genes on both cell lines. Furthermore, the effects of EZH2 protein which belongs to PRC4 protein complex on these epigenetic regulations were also studied.
For this aim we firstly determined the expressions of PRC4 genes with reverse transcriptase (RT)-PCR in both cell lines and our RT-PCR results showed that PRC4 genes, expressed only in embryonic stem/progenitor and cancer cells (12), were expressed in both imatinib-resistant K562/IMA-3 and parental-sensitive K562 cells.
To our knowledge, this is the first report determining the expression levels of EZH2, SUZ12, EED2, and SIRT1 genes in CML cells. Higher EZH2 expression was detected in breast cancer patients especially with metastatic breast cancer, and a correlation was shown with poor prognosis.Citation18 Kuzmichev et al.Citation19 showed that the expressions of PRC4 group proteins were regulated during cancer development and cell differentiations. Overexpression of the EZH2 was studied in lymphomas,Citation20 melanomas,Citation21 renal cell carcinomasCitation22,Citation23 and pancreatic tumor cells.Citation24 We also studied the expression of SUZ12 in both imatinib-resistant K562/IMA-3 and -non-resistant K562 CML cells. In another study, overexpression of the SUZ12 was detected in blastic crisis of CML, as compared with the chronic phase CML patients. Nevertheless, SUZ12 expression was not detected in bone marrow mononuclear cells obtained from a normal donor. For this reason, researchers claimed that SUZ12 could be potentially transformed.Citation25 Expression of SIRT1 was detected in both cell lines in our study. This enzyme belongs to the sirtuin family of histone deacetylases, and has been an important regulator of cell survival in response to various cell stresses.Citation26 It is known that SIRT1 plays a critical role in the initation and development of tumor, drug resistance, cellular senescence and inhibition of apoptosis.Citation27,Citation28 In our study, we also determined SUZ12 expressions in blastic phase of cell lines with enhanced proliferating capacity and resistance to apoptosis. So, we suggest that the expressions of SUZ12 could contribute to the inhibition of apoptotic process.
On the other hand, we examined the epigenetic regulation of BIM and BID pro-apoptotic genes that could play a role in imatinib resistance commonly observed in blastic phase of CML. We focused on BIM and BID pro-apoptotic genes since they may have significant roles in drug resistance, especially in inhibition of apoptosis, partially. In a study reported by Kurado et al.,Citation29 it was found that imatinib caused enhanced BIM expression in K562 and BV173 human leukemia cells. They also showed that BIM played a critical role in imatinib-induced apoptosis. McGarvey et al.Citation30 also found that BIM was able to induce apoptosis in K562 cells.
In our study, we examined the association between BIM and BID genes methylation and PRC4 protein complex in imatinib-resistant K562/IMA-3 and K562 CML cells, and examined whether EZH2 and DNMT1 were bound to the BIM and BID promoter regions and the presence of H3K27me3 modification in these promoters. It was previously demonstrated that H3K27me3 modification caused an accumulation of DNMT1 enzyme in the promoter of genes.Citation31 For interrogation of our hypothesis, first we determined the expression of the PRC4 genes in both imatinib-resistant K562/IMA-3 and K562 cells with conventional PCR. We also determined that EZH2, EED2, SUZ12, and SIRT1 genes were lower expressed and BIM and BID genes were higher expressed in K562/IMA-3 cells as compared with K562 cells.
Results of CHIP experiments revealed that E2H2 and DNMT1 enzymes were bounded to the promoter of the BIM gene in both cell lines. H3K27me3 modification in the promoter of the BIM and BID gene was also detected in both cells while the promoter region of BIM and BID genes was detected as homozygously unmethylated.
E2H2 and DNMT1 enzymes were found to be bounded to the promoter of the BID gene in K562 cells while they were free in K562/IMA-3 cells. H3K27me3 modification in the promoter of the BID gene was determined in both cells. The results of MS-PCR specific to the BID promoter region demonstrated that it was homozygously unmethylated in both cell lines, same as the BIM gene. All these results may suggest the presence of H3K27me3 modification in the promoter regions of BIM and BID genes is not sufficient for methylation of these genes. Consistent with our observations, it was previously shown that BIM was not methylated in K562 cell; however, low levels of expression of Bim protein was determined which may indicate that there may be a different mechanism regulating the expression of Bim protein.Citation32
EZH2 histone methyltransferase-regulated H3K27me3 modification is a hallmark of inactive histone. It was previously demonstrated that H3K27me3 modification is a hallmark of binding of DNMT1 enzyme to the promoter region of the gene in primary colon cancer cells and Caco-2 colon cancer cells.Citation33 Interestingly, McGarvey et al.Citation30 determined that silencing of MLH1, p16, MYT1 and WNT1 genes via DNA methylation do not need the EZH2 enzyme. While an association between gene silencing via PcG proteins and sensitivity to methylation was detected, PcG proteins were not sufficient to explain all CpG methylations observed in cancer. There may be some other factors, rather than the PcG proteins, having roles in DNA methylation.Citation34 Consistent with our resuts, it was previously shown that the promoter regions of the silenced genes which had H3K27me3 modification were unmethylated in HL-60 and OCI-AML3 leukemia cells.Citation35 All these findings indicate that the presence of inactive histone hallmarks like H3K27me3 may sensitize the genes to DNA methylation but they do not always cause de novo DNA methylation.Citation36
Our results suggest that the pro-apoptotic BIM and BID genes are not methyllated imatinib-resistant CML cells. These results may also support the notion that DNA promoter methylation is formed independently from EZH2-H3K27me3.
Funding
This study was supported by TUBITAK-SBAG(Project Number 109S285). This study was partially supported by Turkish Academy of Sciences (TUBA).
References
- Zheng C, Haak M, Brors B, Frank O, Giehl M, Fabarious A: Gene expression profiling of CD33+ cells identifies a molecular signature of chronic myeloid leukemia blast crisis. Leukemia 2006;20:1028–34.
- Cillioni D, Saglio G. CML: a model for targeted therapy. Best Pract Res Clin Haematol. 2009;22:285–94.
- Radich JP. The biology of CML blast crisis. Hematol Am Soc Hematol Educ Program Book, vol 1, 2007;384–91.
- Bixy D, Talpaz M. Mechanism of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia recent therapeutic strategies to overcome resistance. Hematol Am Soc Hematol Educ Program Book, vol 1, 2009;461–7.
- Barnes DJ, Melo JV. Primitive, quiescent and difficult to kill: the role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle. 2006;5:2862–6.
- Baran Y, Ural AU, Gunduz U. Mechanisms of cellular resistance to imatinib in human chronic myeloid leukemia cells. Hematology 2007;12(6):497–503.
- Baran Y, Salas A, Senkal CE, Gunduz U, Bielawski J, Obeid LM, et al. Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J Biol Chem. 2007;282(15):10922–34.
- Smith LT, Otterson GA, Plass C. Unraveling the epigenetic code of cancer for therapy. Trends Genet. 2007;23:449–56.
- Fandy TE, Carraway H, Gore SD. DNA demethylating agents and histone deacetylase inhibitors in hematologic malignancies. Cancer J. 2007;13:40–4.
- Patra SK, Szyf M. DNA methylation-mediated nucleosome dynamics and oncogenic Ras signalling insights from FAS, FAS ligand and RASSF1A. FEBS J. 2008;275:5217–35.
- Martinez AM, Cavalli G. The role of polycomb group proteins in cell cycle regulation. Cell Cycle 2006;5:1189–97.
- Negishi M, Saraya A, Miyagi S, Nagao K, Inagaki Y, Nishikawa M: BMI1 cooperates with Dnmt1-associated protein 1 in gene silencing. Biochem Biophys Res Commun. 2007;353:992–8.
- Kanno R, Janakıraman H, Kanno M. Epigenetic regulator polycomb group protein complexes control cell fate and cancer. Cancer Sci. 2008;99:1077–84.
- Simon JA, Lange CA. Roles of the EZH3 Histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–9.
- Martin C, Zhang Y. The diverse functions of histon lysine methylation. Nat Rev. 2005;6:838–47.
- Besy JD, Carey N. Epigenetic covalent modifications of DNA and chromatin proteins strongly effect gene expression and cellular activity, and epigenetic misregulation occurs in several diseases, especially cancer. Drug Discov Today 2010;15:65–70.
- Pfaff MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:45.
- Ding L, Kleer CG. Enhancer of Zeste 2 as a marker of preneoplastic progression in the breast. Cancer Res. 2006;66:9352–93.
- Kuzmichev A, Margueron R, Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc Natl Acad Sci USA 2005;102:1859–64.
- Garcia EM, Licht JD. Deregulatin of H3K27 methylation in cancer. Nat Genet. 2010;42:100–1.
- McHugh BJ, Fullen DR, Kleer CG. Expression of polycomb group protein EZH2 in nevi and melanoma. J Cutan Pathol. 2007;34:597–600.
- Wagener N, Holland D, Bulkescher J, Mertens IC, Seyler KH, Zentgraf H. The enhancer of zeste homolog 2 gene contributes to cell proliferation and apoptosis resistance in renal cell carcinoma cells. Int J Cancer 2008;123:1545–50.
- Hinz S, Kempkensteffen C, Christoph F, Krause H, Schrader M, Schostak M. Expression parameters of the polycomb group protein BMI1, SUZ12, RING1 and CBX7 in urothelia carcinoma of the bladder and their prognostic revelance. Tumour Biol. 2008;29:323–9.
- Ougolkov AV, Bılım VN, Billadeau DD. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase EZH2. Clin Cancer Res. 2008;14:6790–6.
- Pizzati L, Binato R, Cofre J, Gomes BE, Dobbin J, Hausmann ME. SUZ12’ is a canditate target of the non-canonical WNT pathway in the progression of chronic myeloid leukemia. Genes Chromosomes Cancer 2010;49:107–18.
- Brooks CL, Gu W. How does SIRT1 affect metabolism, senesence and cancer? Nat Rev Cancer 2009;9:123–8.
- Luı T, Iiu PY, Marshall M. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009;69:1702–5.
- Kwon HS, Ott M. The ups and downs of SIRT1. Trends Biochem Sci. 2008;33:517–25.
- Kurado J, Puthalakt H, Cragg MS, Kelly PN, Boullet P, Huang D. Bim and Bad mediate imatinib-induced killing of BcrIAbl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci USA 2006;103:14907–12.
- McGarvey KM, Greene E, Fahrener JA, Jenuwein T, Baylin SB. DNA Methylation and complete transcriptional silencing of cancer genes persist after depletion of EZH2. Cancer Res. 2007;67:5097–102.
- Widschwendter M, Fiegl H, Egle D, Müeller-Holzner E, Spizzo G, Marth C. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–8.
- Jose-Eneriz E, Agirre X, Jimenez-Velasco A, Cordeu L, Martin U, Arqueros U. Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukemia. Eur J Cancer 2009;45:1877–89.
- Schlesnger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–6.
- McCabe MT, Lee EK, Vertino PM. A Multifactorial signature of DNA sequence and polycomb binding predicts aberrant CpG island methylation. Cancer Res. 2009;69:282–90.
- Fiskus F, Wang Y, Sreekumar A, Buickley KM, Shi H, Jillella A. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 2009;114:2733–43.
- Kondo Y. Epigenetic cross-talk between DNA methylation and histone modification in human cancers. Yonsei Med J. 2009;50:455–63.