Abstract
Mixed-lineage acute leukemia (MAL) is characterized as acute leukemia involving acute myeloid cells and lymphoid cells at the same time. It is easily misdiagnosed because of the dual characteristics involving both lymphoid and myeloid cells and has a poor prognosis. We retrospectively analyzed the features and treatment effectiveness in a single center in 40 patients with MAL. The morphology was consistent with acute lymphoblastic leukemia (ALL) (47.5%) or acute myeloid leukemia (AML) (20%) or was inconclusive (32.5%). Twenty-two patients were characterized as B/myeloid, and 18 patients as T/myeloid. Cytogenetics showed t(9;22)/(Ph+) (12.5%) and 11q23/MLL rearrangements (6.25%). The rate of first complete remission for patients undergoing chemotherapy based on the features of both ALL and AML and of either ALL or AML was 71.4 and 42.9%, respectively. The 1-year overall survival rates were 37.5 and 60.0% for chemotherapy and chemotherapy followed by haploidentical hematopoietic stem cell transplantation (HSCT), respectively. The 1-year disease-free survival rates were 25.0 and 50.0% for chemotherapy and chemotherapy followed by HSCT, respectively. These results showed that MAL is confirmed to be a poor-risk disease. The chemotherapy for remission induction should be based on both myeloid cells and lymphoid cells. Transplantation should be performed after the first remission.
Introduction
Mixed-lineage acute leukemias (MALs), also called acute leukemias of ambiguous lineage, represent a heterogeneous category of rare, poorly differentiated acute leukemias that possess characteristics of both lymphoid and myeloid precursor cells.Citation1–Citation8 Divergent morphological and immunophenotypic features may be present throughout a single blast population or may be observed in distinct blast populations in a single MAL patient. MALs represent only 3–5% of acute leukemias, occurring in patients of all ages and comprising several different subtypes. All data indicate that MAL has a poor prognosis,Citation1 and therefore, new methods should be explored to improve the outcome of this disease.
The World Health Organization (WHO) classification has established and published new criteria for the diagnosis of MAL in the recent phase. Briefly, the WHO definition of MAL is based on the expression of strictly specific myeloid (myeloperoxidase (MPO)) and T-lymphoid (cytoplasmic CD3) antigens, the former shown by either cytochemistry or flow cytometry and/or clear evidence of monocytic differentiation. B-cell lineage assignment in MAL relies on the strong expression of CD19 together with another B-cell-associated marker or, in cases with weak CD19, on the expression of at least three B-lineage markers because there is no single antigen strictly specific for B-cells.Citation7 In addition, the WHO recognizes two distinct categories: MAL with the t(v;11q23)/MLL rearrangement and MAL with the t(9;22)(q34;q11)/BCR-ABL1 rearrangement. Whether the biphenotypic acute leukemia cases classified on the basis of the European Group for the Immunological Classification of Leukemias (EGIL) scoring system could have been classified as MAL on the basis of the WHO classification is unclear.
The treatment for MAL patients with acute lymphoblastic leukemia (ALL) or ALL-like regimens has been reported to potentially lead to a higher complete remission (CR) rate than with acute myeloid leukemia (AML) regimens, although this result is controversial.Citation8,Citation9 Zheng et al.Citation9 report that this treatment should be effective for MAL with both ALL and AML features. Whether the treatment for MAL patients should be based on either ALL or AML or both ALL and AML features and, if so, how to combine the chemotherapy drugs and begin the consolidation chemotherapy for the MAL patients is still unclear. Whether the optimal method to treat MAL involves normal chemotherapy or chemotherapy followed by hematopoietic stem cell transplantation (HSCT) also remains unclear. In this study, we retrospectively analyzed the features and effective treatment of 40 patients with MAL in a single center in China.
Patients and methods
Patients
Forty patients diagnosed with MAL between October 2003 and December 2011 in the Department of Hematology, Xinqiao Hospital, Third Military Medical University (China) were entered into this study. Our treatment protocol was approved by the Ethics Committee of The Third Military Medical University, and written informed consent was obtained from all donors, adult patients, and the parents of children <18 years old.
Flow cytometry was used to detect the expression of lineage markers. The diagnosis was performed according to the criteria of EGIL.Citation7 All the patients underwent haploidentical HSCT because of the long waiting period for unrelated doors and there were no available related donors. summarizes the salient patient characteristics.
Table 1. Patient characteristics
Chemotherapy for remission induction
Chemotherapy for remission induction was used with a combined regimen for both AML and ALL based on the morphological features. CVTLP/CVTLP + Ara-c (C, cyclophosphamide; V, vincristine; T, pirarubicin; L, L-asparaginase; P, prednisone; Ara-C, cytarabine) was used for MAL with the morphological features of ALL. MA/TA or MAD/TAD (M, mitoxantrone; A, cytarabine; D, dexamethasone; T, pirarubicin) was used for MAL with the morphological features of AML. MAOD/TAOD (M, mitoxantrone; A, cytarabine; D, dexamethasone; O, vincristine; T, pirarubicin) was used for MAL with the morphological features of both AML and ALL.
Consolidation chemotherapy
Chemotherapy for consolidation chemotherapy was also used with a combined regimen for both AML and ALL based on the morphological features. CTOD + Ara-c (C, cyclophosphamide; V, vincristine; T, pirarubicin; D, dexamethasone; Ara-C, cytarabine) with normal doses + media-high methotrexate (3 g/m2) was used for MAL with the morphological features of ALL. MD/TD with normal doses + media-high Ara-c (M, mitoxantrone; D, dexamethasone; T, pirarubicin; Ara-c,: cytarabine) was used for MAL with the morphological features of AML. MAOD with normal doses (M, mitoxantrone; A, cytarabine; V, vincristine; D, dexamethasone) was used for MAL with the morphological features of both AML and ALL.
HSCT donors
All donors were relatives of the patients, such as the father, mother, brother, or sister. All donors had three mismatch loci with HLA-A, B, and DR. The degree of the locus mismatch conformed to the Chinese consensus on immunogenetic donor searches for full mismatch transplantation. Pre-transplantation lymphocyte cross-matches with the patients’ sera, and donor cells were negative in all cases.
Harvest procedures
The mobilization and harvest of stem cells was performed as previously reported.Citation10
Treatment regimens
Two conditioning regimens were used: (i) Ara-c (at a dose of 3.0 g/m2) for 3 days combined with intravenous (IV) cyclophosphamide at 45 mg/kg/day for 2 consecutive days and total body irradiation (TBI) daily for 2 consecutive days (9.0–9.5 Gy total dose); and (ii) busulfan (0.4 mg/kg/day) combined with cyclophosphamide (1.8 g/m2) and Ara-c (2.0 g/m2 q12 hours) for 2 consecutive days. Regimen (i) was performed on two patients and regimen (ii) on the eight remaining patients. All patients received cytotoxic drugs and TBI at a planned dose (100% ± 5%). Granulocyte-colony stimulating factor (G-CSF) at a dose of 5 µg/kg was started 24 hours after transplantation and continued until engraftment. Graft-versus-host disease (GVHD) prophylaxis was performed according to our previous study.Citation11
Evaluation and definitions
CD34+ cells were identified in the leukapheresis product by flow cytometry as previously described.Citation12 Neutrophil engraftment was defined as the first of 3 consecutive days with an absolute neutrophil count (ANC) higher than 0.5 × 109/l. Platelet engraftment was defined as the first of 7 consecutive days on which the platelet count exceeded 20 × 109/l without transfusions. Acute and chronic GVHD were graded according to standard criteria. Treatment-related mortality was defined as death in the absence of relapse. Relapse was diagnosed from bone marrow specimens taken on day +30, day +100, or when clinically indicated. The disease-free survival (DFS) was calculated from the transplantation to the end of follow-up.
Statistical analysis
The statistical evaluation was performed using Student's t-test with SPSS software (version 10.0, Chicago, IL, USA). The survival curve was derived using a Kaplan–Meier analysis, and the survival rate and mortality were analyzed using log-rank and Breslow tests, respectively. A significance level of P < 0.05 was used.
Results
Morphology
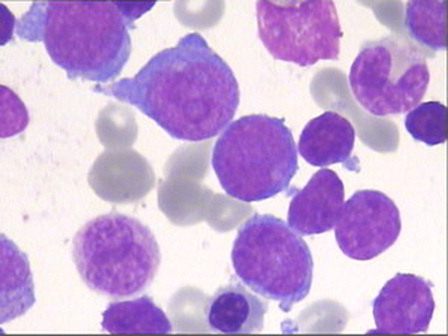
A review of peripheral blood and bone marrow smears for morphology and cytochemistry was performed in 40 cases. According to the FAB criteria, 19 (47.5%) cases displayed an ALL morphology. Eight (20%) cases were classified as AML, namely M1 and M5 and rarely M2 or M4 according to the FAB; no single case was M3, M6, or M7 or had a dysplastic background. The remaining 13 (32.5%) cases had a dual population of small and large blasts difficult to classify by morphology and were categorized as undifferentiated acute leukemia ().
Figure 1. May–Grunwald–Giemsa-stained BM smear showing a mixed-cell population of large and small blasts.
Flow cytometric immunophenotyping
Immunophenotyping showed that 22 (55.0%) cases had a B-lymphoid + myeloid immunophenotype (B + My), 18 (45%) a T-lymphoid + myeloid immunophenotype (T + My). There were no significant differences between the B + My and T + My immunophenotypes for age, sex, and morphology.
HLA-DR was strongly positive in 32 (86.5%) of 37 cases, and CD34 in 17 (45.9%) of 37 cases. The expression results of other antigens are described in the next section according to the myeloid and T- and B-lymphoid commitment.
Myeloid commitment in cases with a B + My and T + My immunophenotype
The MoAb anti-MPO was expressed in at least 10% of blasts in 17 (45.9%) of 37 cases. Therefore, there was evidence of myeloid commitment in all cases. There was a variable proportion of blasts (10–95%) coexpressing anti-MPO and lymphoid markers in all but the eight patients in whom only MPO cytochemistry was available and in whom double staining could not be performed.
In our cohort of patients, CD13 was positive in 18 (48.6%) of 37 cases, and CD33 in 16 (43.2%) of 37 cases. Six cases expressed MPO together with CD33 and/or CD13. The expression of other myelo-/monocyte-associated markers was as follows: CD14 in 10 (27.0%) cases and CD117 in 4 (10.8%) cases.
T-lymphoid commitment in cases with a T + My immunophenotype
Cytoplasmic CD3 (cytCD3) was expressed in all 11 cases. In five cases, the blasts coexpressed cytCD3 and anti-MPO. CD5 was positive in 10–95% of blasts from 10 (62.5%) of 16 cases, and CD7 in 12–98% of blasts from 11 (68.8%) of 16 cases tested. The expression of other lymphoid-lineage-associated markers was as follows: CD10 in 2 (12.5%) of 16 cases, and CD79a in 1 (6.25%) of 16 cases.
B-lymphoid commitment in cases with a B + My immunophenotype
CD19 was expressed in 16 (76.2%) of 21 cases. In all of these cases, the blasts were positive for CD10, cytCD22, and/or cytCD79a. The expression of other lymphoid markers in this entire group was as follows: CD10 in 12 (57.1%) of 21 cases, CD20 in 9 (42.9%) of 21 cases, and cytCD22 in 4 (19.0%) of 21 cases.
The WHO 2008 criteria
We analyzed whether the MAL cases included here according to the EGIL scoring system could have been classified as mixed-phenotype acute leukemia (MPAL) on the basis of the WHO 2008 criteria. Seventeen of 21 cases with B-lymphoid commitment scored 2.5 or greater for this lineage, and 11 cases with T-lymphoid commitment scored 2.5 or greater. Regarding the myeloid commitment, all but five cases scored more than 2. Cells from the 17 cases that scored only 2 had evidence of myeloid commitment or lymphoid commitment by both immunostaining and cytochemistry, but were negative for MPO or CD3 and/or not all the myeloid/lymphoid-associated markers were tested; therefore, they would not have been classified as MPAL according to the WHO 2008 criteria.
Cytogenetics
Of the 16 patients in whom cytogenetic information was available, 2 (12.5%) had the t(9;22)(q34;q11) arrangement (Ph) confirmed by fluorescence in situ hybridization, and 1 (6.25%) case had translocations involving the 11q23 breakpoint (MLL gene) with t(9;11)(p22;q23).
Chemotherapy for remission induction
Information on the response to the first-line treatment was available for 34 patients; six patients did not receive treatment and were discharged. Out of these 34 patients, 15 patients had the morphological features of ALL, 6 patients had the morphological features of AML, and the other 13 patients had the morphological features of both AML and ALL. With the available data from 14 patients with the morphological features of ALL, 12 patients reached CR after remission induction, 1 patient reached partial remission (PR), and the remaining 1 patient was non-remissive (NR). Of the six patients with the morphological features of AML, three reached CR after remission induction, two patients reached PR, and the remaining one patient was NR. Of the 10 patients with the morphological features of both AML and ALL, 5 patients reached CR after remission induction, 3 patients reached PR, and the remaining 2 patients were NR. The total percentage of patients who reached CR was 66.7%.
Twenty-three patients underwent chemotherapy based on the features of both ALL and AML. Of these, 17 patients reached CR, and the rate of CR was 73.9%. Seven patients underwent chemotherapy based on the features of ALL or AML. Of these, three patients reached CR, and the rate of CR was only 42.9%. The difference between the CR rates of these therapies was significant (P<0.05).
Consolidation chemotherapy
The consolidation chemotherapy was also based on the morphological features. Of the eight patients with PR and NR, only one patient was available for consolidation chemotherapy. This patient reached CR after one session of consolidation chemotherapy. One patient who reached CR was released from treatment during the first instance of consolidation chemotherapy. The other 18 patients showed continuous CR.
Engraftment
Ten patients who underwent HSCT manifested stable neutrophil and platelet engraftment; there was no late graft failure. The median days that the white blood cell levels achieved an ANC>0.5 × 109/l and platelet levels reached >20 × 109/l were +14.5 (10–25) and +14 (13–25) post-transplantation, respectively.
Acute and chronic GVHD
Six of ten patients showed no signs of acute GVHD. Three patients developed a slight acute GVHD (overall grade II), and one patient experienced an overall grade III acute GVHD. Chronic GVHD was not observed in any patient.
Treatment-related mortality, relapse, overall survival, and follow-up
Treatment-related results were collected from 18 patients who underwent at least one treatment with consolidation chemotherapy or chemotherapy followed by haploidentical HSCT; eight patients underwent chemotherapy at least twice. According to their willingness, 10 patients underwent chemotherapy followed by haploidentical HSCT after receiving two cycles of consolidation chemotherapy, and the disease continuously maintained CR. There were no available related/unrelated HLA-matched donors. For all patients, the median follow-up duration was 10 (1–78) months. The 1-year overall survival (OS) and DFS were 40.0 and 32.0%, respectively.
For patients with normal chemotherapy, the median follow-up duration was 9 (3–78) months. Five of these patients died, and the median survival time was 12 months. For patients who underwent HSCT, the average age was 28 years (range 9–48), and the treatment-related mortality was zero. The median follow-up duration was 11.5 (6–56) months. Four patients died (one died of a fungal infection 6 months post-transplantation, and three died after relapse), and the median survival time was 8.5 (6–20) months. The other patients survived through the study period. The 1-year OS rates were 37.5 and 60.0% for chemotherapy and chemotherapy followed by HSCT, respectively (P > 0.05 for the log-rank test and P < 0.05 for the Breslow test). The 1-year DFS rates were 25.0 and 50.0% for chemotherapy and chemotherapy followed by HSCT, respectively (P < 0.05 for both the log-rank and Breslow tests).
Discussion
MAL is an extremely rare form of leukemia,Citation13 and the mechanism of its pathogenesis remains unclear. It has been reported that translocation, rearrangement, and the loss of some genes may be involved in the development of this rare disease.Citation14 Extensive data on its response to therapy and its clinical outcome are still not available. However, all reports in the literature indicate a dire outcome.Citation15 There are no uniform criteria for treating this disease as ALL or AML or by employing an approach that combines effective drugs for ALL and AML followed by HSCT after CR.Citation16 In this study, we retrospectively analyzed the effects of treating patients with MAL with a combined regimen for both ALL and AML followed by haploidentical HSCT and achieved a favorable outcome.
Information on the characteristic features of MAL is very limited because of the rarity of these leukemias and because of the new criteria established for the definition of MAL. The diagnostic criteria for MAL until now were based on the scoring system proposed by EGIL that was adopted by the WHO 2001 classification. Thus, a substantial number of publications refer to data on patients with miscellaneous diagnoses including true MAL, and some cases that would not fulfill the present WHO criteria. Our results showed that only a minority of cases included here would not have been classified as MPAL according to the 2008 WHO criteria.
MAL represents 8% of leukemia cases and has a poor prognosis with a 4-year survival rate of only 8%.Citation4 Killick et al.Citation5 described 20 patients with MAL (8 children and 12 adults) and reported that CR was achieved in 70% of patients, although the probability of survival at 2 years was only 39%. However, all eight children with this type of leukemia achieved CR (five after ALL induction, two after AML induction, and one after a switch to AML induction after a poor response to ALL induction therapy). Six of these patients survived and were in remission at 2 years of follow-up. A study of seven patients with MAL that expressed T-lymphoid and myeloid markers showed that only two of seven patients entered CR after ALL induction therapy, but four of the remaining five attained CR after receiving AML therapy.Citation3 In another single-center study involving 31 adult patients with MAL, the CR rates were 78% for patients who received lymphoid-directed therapy and 57% for those who received myeloid therapy, whereas the OS at 2 years was 60%.Citation17 A recent study proposed that remission induction for MAL patients with ALL or ALL-like regimens may lead to higher CR rates than those with AML regimens; however, this approach is controversial.Citation8,Citation9 Wang and XuCitation8 have hypothesized that a combined regimen for both AML and ALL might be the best choice for the induction therapy for MAL, but whether the combination is effective and how to combine these approaches are still unknown. We believe that the morphology is a manifestation of both cytogenetics and molecular biology. In this study, we treated patients with MAL with a combined regimen for both AML and ALL for the induction chemotherapy, and a high CR was reached. However, because these cases occurred over a period of 8 years, the differences in the support care and chemotherapy drugs during different phases may have influenced the outcome, which should be further explored with more cases and in multiple centers.
Haploidentical HSCT is an interesting alternative when no sibling or unrelated matched donors are available. Although T-cell-depleted allo-HSCT from haploidentical relatives is a feasible option for patients who need an allograft but lack a compatible donor, the graft failure rate is high and is mainly mediated by host alloreactive T-cells that survive the preparative regimen. The recipients of T-cell-depleted allo-HSCT from disparate relatives are also exposed to an increased risk of life-threatening infections, especially viral infections, due to the delay in hematopoiesis and adaptive immunity.Citation18 Allo-HSCT with T-cells also has a risk of complications, such as serious GVHD and infections.Citation19 Compared with G-CSF-mobilized peripheral blood grafts (G-PB), the use of G-CSF-primed bone marrow (G-BM) resulted in comparable engraftment, reduced the severity of acute GVHD, and resulted in less subsequent chronic GVHD; moreover, G-BM transplants produced even fewer cases of chronic GVHD than steady-state bone marrow transplantation.Citation20 To reduce the incidence of GVHD while retaining a GVL effect in allo-HSCT, especially in patients without matched donors, a clinical study showed that the incidence of GVHD was not higher than that of bone marrow transplantation and G-PB transplantation in patients with HLA-matched donors using G-BM plus G-PB without T-cell depletion ex vivo on haploidentical transplantation for patients with malignant hematological disease.Citation12 In this study, we performed haploidentical HSCT with the combination of G-PB and G-BM without T-cell depletion ex vivo. The results showed that the acute GVHD was not serious and that no chronic GVHD was observed, and improved results were achieved compared with the standard chemotherapy.
Our results indicate that a combined regimen for both ALL and AML for the induction and consolidation chemotherapy followed by haploidentical HSCT is a good method to treat MAL. However, the long-term survival of these patients should be studied in a multicenter study with a large number of cases.
References
- Weir EG, Li Ansari-Lari M, Batista DA, Griggin CA, Fuller S, Smith BD, et al. Acute bilineal leukemia: a rare disease with poor outcome. Leukemia. 2007;21:2264–70.
- Rubnitz JE, Onciu M, Pounds S, Shurtleff S, Cao X, Raimondi SC, et al. Acute mixed lineage leukemia in children: the experience of St Jude Children's Research Hospital. Blood. 2009;113:5083–9.
- Rubio MT, Dhedin N, Boucheix C, Bourhis JH, Reman O, Boiron JM, et al. Adult T-biphenotypic acute leukaemia: clinical and biological features and outcome. Br J Haematol. 2003;123:842–9.
- Legrand O, Perrot JY, Simonin G, Baudard M, Cadiou M, Blanc C, et al. Adult biphenotypic acute leukaemia: an entity with poor prognosis which is related to unfavourable cytogenetics and P-glycoprotein over-expression. Br J Haematol. 1998;100:147–55.
- Killick S, Matutes E, Powles RL, Hamblin M, Swansbury J, Treleaven JG, et al. Outcome of biphenotypic acute leukemia. Haematologica. 1999;84:699–706.
- Aribi A, Bueso-Ramos C, Estey E, Estrov Z, O'Brien S, Giles F, et al. Biphenotypic acute leukaemia: a case series. Br J Haematol. 2007;138:213–6.
- Matutes E, Pickl WF, van't Veer M, Morilla R, Swansbury J, Strobl H, et al. Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood. 2011;117:3163–71.
- Wang J, Xu X. What is the optimal treatment for biphenotypic acute leukemia? Authors’ reply. Haematologica 2009;94:1780.
- Zheng C, Wu J, Liu X, Ding K, Cai X, Zhu W. What is the optimal treatment for biphenotypic acute leukemia? Haematologica. 2009;94:1778–80.
- Zhang C, Chen XH, Zhang X, Gao L, Gao L, Kong PY, et al. Stem cell collection in unmanipulated HLA-haploidentical/mismatched related transplantation with combined granulocyte-colony stimulating factor-mobilized blood and bone marrow for patients with hematologic malignancies: the impact of donor characteristics and procedural settings. Transfus Med. 2010;20:169–77.
- Zhang C, Chen XH, Zhang X, Gao L, Gao L, Kong PY, et al. Mobilization of peripheral blood stem cells for autologous transplantation patients with hematologic malignancies: influence of disease, mobilization method, age and sex. Transfus Apher Sci. 2008;39:21–8.
- Chen X, Zhang C, Zhang X, Gao L, Gao L, Kong PY, et al. Role of antithymocyte globulin and granulocyte-colony stimulating factor-mobilized bone marrow in allogeneic transplantation for patients with hematologic malignancies. Biol Blood Marrow Transplant. 2009;15:266–73.
- Downing JR, Shannon KM. Acute leukemia: a pediatric perspective. Cancer Cell. 2002;2:437–45.
- Wu SQ, Kuo J, Chen XR, Chen SA, Quinn JJ. Translocation (6;14) in childhood acute mixed lineage leukemia. Cancer Genet Cytogenet. 2003;141:178–9.
- Daser A, Rabbitts TH. The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin Cancer Biol. 2005;15:175–88.
- Aglietta M, De Vincentiis A, Lanata L, Lanza F, Lemoli RM, Menichella G, et al. Peripheral blood stem cells in acute myeloid leukemia: biology and clinical applications. Haematologica. 1996;81:77–92.
- Handretinger R, Lang P, Klingebiel T, Schumm M, Neu S, Geiselhart A, et al. Megadose transplantation of purified peripheral blood CD34+ progenitor cells from HLA-mismatched parental donors in children. Bone Marrow Transplant. 2001;27:777–831.
- Schattenberg AV, Dolstra H. Cellular adoptive immunotherapy after allogeneic stem cell transplantation. Curr Opin Oncol. 2005;17:617–21.
- Zhang C, Zhang X, Chen XH, Gao L, Gao L, Kong PY, et al. Factors influencing engraftment in HLA-haploidentical/mismatch related transplantation with combined granulocyte-colony stimulating factor-mobilized peripheral blood and bone marrow for patients with leukemia. Transfus Apher Sci. 2011;44:249–55.
- Huang XJ, Han W, Xu LP, Chen YH, Liu DH, Lu J, et al. A novel approach to human leukocyte antigen-mismatched transplantation in patients with malignant hematological disease. Chin Med J. 2004;117:1778–85.