
Abstract
Background
The group of unstable hemoglobins are associated with congenital non-spherocytic hemolytic anemia due to instability of the hemoglobin molecule. They often lead to formation of the characteristic inclusion bodies or Heinz bodies.
Aim
To identity the cause of mild anemia, reticulocytosis, and hepatosplenomegly in a case of non-spherocytic hemolytic anemia.
Materials and methods
A 34-year-old female patient originating from Maharashtra, western India presented with mild anemia and jaundice which had persisted since childhood. Investigations included a complete blood count, screening for red cell membrane protein defects, Hb analysis by high-performance liquid chromatography (HPLC) and cellulose acetate electrophoresis (pH 8.9), heat instability test and DNA sequencing.
Results
Hemoglobin analysis by HPLC showed an abnormal peak in the Hb C window (9.8%) with a retention time of 4.90 minutes. Cellulose acetate electrophoresis (pH 8.9) showed a slow moving band (6.15%) between Hb A2 and Hb S. The heat instability test was positive. DNA analysis of α globin genes showed absence of both deletional and non- deletional α thalassemia. DNA sequencing of the β globin gene revealed heterozygosity for a mutation at codon 98 [GTG → ATG, Val → Met], which gives rise to Hb-Koln.
Conclusion
Hb Koln is the commonest unstable Hb variant reported from many populations in the world. However, this is the first report of this unstable Hb variant from India.
Introduction
The group of unstable hemoglobins are associated with congenital non-spherocytic hemolytic anemia due to instability of the hemoglobin molecule. They often lead to formation of the characteristic inclusion bodies or Heinz bodies. The clinical presentation of patients varies widely from an asymptomatic or mildly anemic condition to a severe disorder. The severity can be largely attributed to the underlying molecular defect. More than 100 different unstable hemoglobins leading to hemolytic anemia have been described so far. Hemoglobin Koln (Hb Koln) is the most common unstable hemoglobin variant with a dominant inheritance pattern. It results from a replacement of valine by methionine at β98 (FG5) causing increased oxygen affinity and instability possibly due to heme depletion.Citation1,Citation2 Here we report the first case of Hb Koln in an Indian patient referred for diagnosis of non-spherocytic hemolytic anemia.
Materials and methods
The proband was a 34-year-old female born of a non-consanguineous marriage originating from Sholapur district in the state of Maharashtra in western India. She had a history of mild anemia and jaundice since childhood but had no history of receiving any blood transfusions. There was no family history of chronic anemia or jaundice. She presented with mild anemia, reticulocytosis, and mild hepatosplenomegaly.
Red cell indices were measured on an automated cell counter (Sysmex K-1000). Peripheral smear examination and reticulocyte count were done by standard methods. Eosin-5-maleimide dye test using flow cytometry was used to screen for red cell membrane protein defects.Citation3 Hemoglobin analysis was done using cation exchange high-performance liquid chromatography (HPLC) on the Variant Hemoglobin Testing System (BioRad Laboratories, Hercules, CA, USA) and by cellulose acetate electrophoresis (pH 8.9). Heat and isopropanol instability tests were undertaken to screen for unstable hemoglobins. Screening for 8 common α thalassemia deletions (−α3.7, −α4.2, –SEA,–SA,–FIL,–MED,–THAI,–20.5) was done using multiplex PCR.Citation4 DNA Sequencing of the α and β globin genes was done using the ABI Prism 310 Sequencer (Applied Biosystems, Foster City, CA, USA) to characterize the hemoglobin variant.
Results
The peripheral smear showed hypochromia, target cells and a reduction in the number of platelets. The hematological and molecular findings in the patient are summarized in . The proband had a hemoglobin level of 11.9 g/dl with a reticulocyte count of 9.0% and showed the absence of Heinz bodies. She had reduced platelet counts (119 × 103/μl) with a normal prothrobin time. Flow cytometric analysis of red cell membrane proteins by using eosin-5-maleimide dye showed the absence of red cell membrane protein defects. Hemoglobin analysis by HPLC showed an abnormal peak in the Hb C window – 9.8% with a retention time of 4.90 minutes (). Cellulose acetate electrophoresis (pH 8.9) showed a slow moving band (6.15%) between Hb A2 and Hb S.
Figure 1. Cation-exchange HPLC analysis on the Variant Hb Testing System; HPLC analysis showed an abnormal peak in the C window – 9.8% with a retention time of 4.90 minutes.
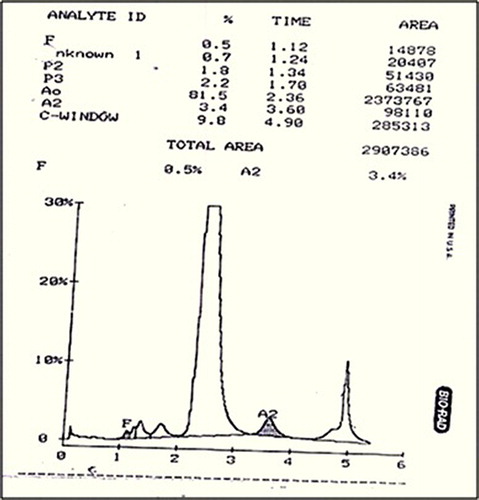
Table 1. Hematological and molecular data of the proband
DNA analysis of the α globin genes showed absence of both deletional and non-deletional α thalassemia. DNA sequencing of the β globin gene revealed heterozygosity for a mutation at codon 98 [GTG → ATG, Val → Met], which gives rise to Hb-Koln ().
Figure 2. DNA sequencing electropherogram of the proband showing the Hb Koln mutation [β98 (FG5) [GTG → ATG, Val → Met]. The arrow indicates the position of the nucleotide substitution GTG (valine) > ATG (methionine).
![Figure 2. DNA sequencing electropherogram of the proband showing the Hb Koln mutation [β98 (FG5) [GTG → ATG, Val → Met]. The arrow indicates the position of the nucleotide substitution GTG (valine) > ATG (methionine).](/cms/asset/652e31b8-93e1-4309-863b-13b28af2a377/yhem_a_11735004_f0002_b.jpg)
Discussion
One hundred and forty-five unstable hemoglobins have been identified so far (http://globin.bx.psu.edu/cgi-bin/hbvar/counter). About 100 of these variants lead to hemolysis or abnormal oxygen affinity while the other variants have no hematological abnormalities but show instability in the in vitro test. The unstable hemoglobin variants have a variable phenotype depending on the position of the mutation and whether they disturb the structure of the globin chain or hemoglobin molecule.Citation5 Hb Koln is the most common of the unstable hemoglobin variants and it was first described by Pribilla in 1962.Citation6 The structural abnormality of the variant was later demonstrated by Carrell et al. in 1966.Citation7 One of the main characteristics of unstable hemoglobins is the presence of Heinz bodies. The severity of the anemia is related to the degree of the resulting hemoglobin instability. Numerous Heinz bodies usually appear in the peripheral blood after splenectomy. In the present case, we did not find any Heinz bodies in the red cells possibly due to the active function of the patient's spleen. There are few Hb variants like Hb O Arab, Hb Constant Spring, Hb O Indonesia, Hb Agenogi, and Hb Siriraj which elute in the Hb C window.Citation8,Citation9 The α chain variants usually have a lower percentage of the abnormal hemoglobin compared to the β chain variants. In view of the low percentage of this variant (9.8%), we first sequenced the α globin gene which was normal. Sequencing of the β globin gene thereafter, led to the identification of Hb Koln. Our patient showed mild thrombocytopenia. A similar finding has been observed in other patients of Hb Koln earlier.Citation10,Citation11 The decrease in platelet count in our patient was most likely due to splenic sequestration of platelets. This is supported by the observation of Pedersen et al., in their hypersplenic patient where thrombocytopenia was corrected after splenectomy.Citation10
Hemoglobin Koln has been previously reported in persons of Northern European ancestryCitation6,Citation7,Citation11 and in Malay,Citation12 Jewish,Citation13 American Blacks,Citation10 Japanese,Citation14 Czechoslovakian,Citation15 and Korean patients.Citation16 This hemoglobinopathy has a widespread distribution although it is being reported from India for the first time.
Conclusion
Hb Koln is the commonest unstable Hb variant reported from many populations in the world. However, this is the first report of this unstable Hb variant from India.
Acknowledgment
This work was supported by the Indian Council of Medical Research.
References
- Horst J, Oehme R, Kohne E. Hemoglobin Koln: direct analysis of the gene mutation by synthetic DNA probes. Blood 1986;68:1175–7.
- Miller DR, Weed RI, Stamatoyannopoulos G, Yoshida A. Hemoglobin Koln disease occurring as a fresh mutation: erythrocyte metabolism and survival. Blood 1971;38:715–29.
- Kedar PS, Colah RB, Kulkarni S, Ghosh K, Mohanty D. Experience with eosin-5′-maleimide as a diagnostic tool for red cell membrane cytoskeleton disorders. Clin Lab Haematol. 2003;25:373–6.
- Tan A, Quah T, Low P, Chang S. A rapid and reliable 7 deletion multiplex polymerase chain reaction assay for α thalassemia. Blood 2001;98:250–1.
- Naito Y, Takahashi T, Matsunashi T, Harano K, Harano T. Hb Nishinomiya [Leu-Gly-inserted between codons 69(E13) and 70(E14) of beta]: a novel unstable hemoglobin with reduced oxygen affinity found in a patient with spherocytic hemolysis. Int J Hematol. 2002;76:146–54.
- Pribilla W. Thalasssaemia-ahnliche Erkrankung mit neuem minor-Hamoglobin (Hb Koln). In: Lehmann H, Betke K (eds.) Haemoglobin-colloquium. Wien, Stuttgart: Thieme; 1962. pp. 73–4.
- Carrell RW, Lehmann H, Hutchison HS. Hemoglobin Koln (b98 Valine-Methionine). An unstable protein causing inclusion body anemia. Nature 1966;210:915–916
- Riou J, Godart C, Hurtrel D, Mathis M, Bimet C, Bardakdjian-Michau J, et al. Cation-exchange HPLC evaluated for presumptive identification of hemoglobin variants. Clin Chem. 1997;43(1):34–9.
- Colah RB, Surve R, Sawant P, D'Souza E, Italia K, Phanasgaonkar S, et al. HPLC Studies in Hemoglobinopathies. Indian J Pediatr. 2007;74:657–64.
- Pedersen PR, McCurdy R, Wrightstone RN, Wilson JB, Smith LL, Huisman THJ. Hemoglobin Koln in a Black: Pre-and post-splenectomy red cell survival (DF 32P and 51Cr) and the pathogenesis of hemoglobin instability. Blood 1973;42:771–81.
- Hutchison HE, Pinkerton PH, Waters P, Douglas AS, Lehmann H, Beale D. Hereditary Heinz-body anemia, thrombocytopenia, and haemoglobinopathy (Hb Koln) in a Glasgow family. Br Med J. 1964;2:1099–103.
- Eng LL, Lopez CG, Eapen JS, Eravelly J, Wiltshire BG, Lehmann H. Unstable Haemoglobin Koln disease in members of a Malay family. J Med Genet. 1972;9:340–3.
- Hallen J, Charlesworth D, Lehmann H. Haemoglobin Koln in a Jewish family. Acta Med Scand. 1972;191:177–80.
- Ohba Y, Miyaji T, Shibata S. Identical substitution in Hb Ube-I and Hb Koln. Nat. New Biol. 1973;243:205–7.
- Indrak K, Brabec V, Wilson JB, Webber BB, Huisman THJ. Hb Koln or α2 β2 98 (FG5) Val → Met in a Czechoslovakian family. Hemoglobin 1991;15:133–5.
- Chang YH, Hur M, Lee DS, Park SS, Kim BK, Park S, et al. The first case of Hb Koln [β98 (FG5) Val → Met] in Korea. Hemoglobin 1999;3:287–9.