
Abstract
Objective and Importance
Post-transplant lymphoproliferative disorder (PTLD) is a severe complication after allogeneic hematopoietic stem cell transplantation (allo-HSCT) associated with Epstein–Barr virus (EBV).
Clinical presentations
Among 263 individuals treated with allo-HSCT for severe aplastic anemia, pure white cell aplasia, T-prolymphocytic leukemia, and relapsed Hodgkin lymphoma, we diagnosed EBV-PTLD in 5 patients. Median age was 29 years (range 19–70 years) and four of five patients were EBV-seropositive prior to HSCT. All five had unrelated EBV-positive donors. In all cases, PTLD occurred within the first year post-transplant (median 4 months).
Intervention
There were two rapidly fatal courses with extensive organ involvement. Both patients showed lymphopenia and thrombocytopenia. In contrast, the three surviving patients had higher lymphocytes and normal platelet counts, while PTLD was restricted to one site and resolved after 2–4 cycles of rituximab.
Conclusion
In this case series courses of PTLD showed substantial diversity.
Introduction
Post-transplant lymphoproliferative disorder (PTLD) is a severe complication after allogeneic hematopoietic stem cell transplantation (HSCT).Citation1 PTLD is usually associated with a reactivation of latent Epstein–Barr virus (EBV) or rarely also with de novo infection leading to increased proliferation of B-lymphocytes driven by EBV.Citation2–Citation6 In the setting of incomplete immune reconstitution after HSCT, EBV-driven B-cell proliferation is insufficiently controlled by T-cells and potentially gives rise to post-transplant lymphoma. Particularly, the early forms of PTLD occurring within the first year after HSCT may show a rapid course with considerable morbidity and mortality. Studies by the Center for International Blood and Marrow Transplant Research revealed T-cell depletion, treatment with anti-T-cell antibodies such as anti-T-cell globulin (ATG) or alemtuzumab, and unrelated stem cell donors as important risk factors to develop PTLD.Citation1,Citation7 We identified 5 cases among 263 patients undergoing allogeneic HSCT, which show that the severity of PTLD course can be highly diverse.
Patients and methods
The courses and outcomes of EBV-associated PTLD were analyzed at our center over a period of 4.5 years between January 2007 and June 2011. In all cases of clinically manifest PTLD, we evaluated peripheral blood counts, EBV viral load and organ involvement, as well as lymph node histology when available. Conditioning regimens, Graft versus host disease (GvHD) prophylaxis, therapies preceding the transplant and treatments after allogeneic HSCT were assessed. We performed no monitoring of EBV DNA viral load routinely after HSCT. Only in the suspicion of a lymphoma, EBV viral load was determined. All patients gave informed consent to the analysis of their data in accordance to the declaration of Helsinki.
Results
Over a period of 4.5 years (January 2007 to June 2011) 5 patients developed symptomatic PTLD among 263 individuals undergoing allogeneic HSCT. This corresponds to a prevalence of 1.9% at our center. Underlying disorders were severe aplastic anemia (SAA) (, patients 1 and 3), pure white cell aplasia (, patient 5), T-prolymphocytic leukemia (, patient 2), and relapsed Hodgkin lymphoma (, patient 4). All five patients had onset of PTLD within the first year post-transplant with a peak at 4 months. All five patients had an unrelated donor. Conditioning chemotherapy included ATG in two patients (, patients 1 and 5), fludarabine in another two patients (, patients 2 and 3), and combination of fludarabine and ATG in one patient (, patient 4). Prior to starting conditioning treatment for allogeneic HSCT two patients had undergone T-cell depleting therapies such as alemtuzumab or ATG (, patients 2 and 3). One patient rejected the allogeneic transplant and was subsequently treated by alemtuzumab (, patient 5).
Table 1. Patient characteristics
Two of the five patients had an aggressive and rapidly fatal course with extensive organ involvement and were refractory to intensive therapy (, patients 1 and 2). In contrast, patients 3–5 had milder PTLD restricted to one site and responsive to 2–4 cycles of rituximab. Both patients with extensive and fatal disease showed thrombocytopenia at the time of PTLD diagnosis, whereas the three patients with a more benign course had normal platelet counts (). Lymphopenia at the time of PTLD diagnosis was present in three patients, while two of them died of the PTLD (). C-reactive protein level was higher in the patients with a fatal course (116.8 and 157.8 mg/l in patients 1 and 2) than in the surviving patients (9.8, 102.3, and 3.6 mg/l in patients 3–5). Higher EBV viral loads did not constantly associate with more severe courses (). One of the patients with fatal outcome (, patient 1) had primary EBV infection, and therefore no residual immunological memory for EBV. The EBV viral loads in the course of the five patients are shown in .
Table 2. Characteristics of PTLD, treatment, and outcome
Table 3. EVB viral loads in the course and therapy
Case presentations
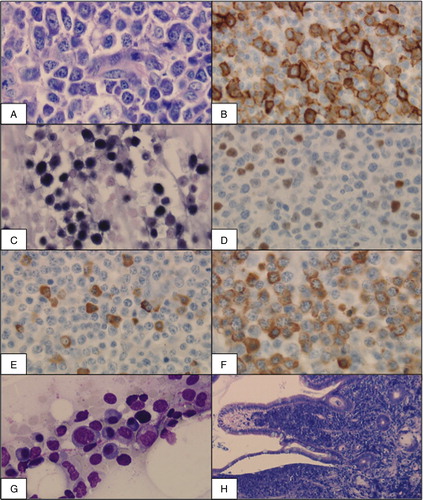
Patient 1 was a 19-year-old EBV-seronegative man with SAA. He directly underwent allogeneic bone marrow transplantation (BMT) from a fully matched, EBV-seropositive unrelated donor after conditioning with cyclophosphamide and ATG (). Two months after BMT severe primary EBV infection was diagnosed with gross cervical, axillary, mediastinal, and mesenterial lymphadenopathy, swollen tonsils and an EBV viral load of 4.400.000 genome equivalents per milliliter (GEq/ml) (). In addition pancytopenia and upper gastrointestinal bleeding developed. Histological analysis of lymph nodes (A–D) and intestinal tumors of up to 3.5 cm diameter (F) showed infiltration by an Epstein–Barr encoded RNA (EBER)-positive plasmablastic lymphoma with expression of CD20 in 30% of the cells and very high proliferation activity (Ki-67 99%). Bone marrow was infiltrated by an early lesion type of PTLD, i.e. plasmacytic hyperplasia (E). Immunosuppression with cyclosporine A at a dose of 325 mg/day at the time of PTLD diagnosis was stopped. Treatment with rituximab (375 mg/m2/week) initially led to clinical regression and reduced EBV viral load. After the third application of rituximab, the lymphoma progressed and extensive lymphomatous infiltration of the lungs rapidly developed. Histological analysis showed a nearly complete loss of CD20 positivity (1–2%). Chemotherapy with R-CHOP as well as intensified treatment with etoposide and cytarabine was ineffective. The patient died from multi-organ failure 3 months after BMT. Donor lymphocyte infusions (DLIs) as well as EBV-specific donor cytotoxic T-lymphocytes (CTLs) were in preparation, but not available in time.
Figure 1. Histological analysis in patient 1 showing lymph node (A–F), bone marrow (G), and intestinal mucosa (H). (A) Cervical lymph node (Giemsa, 400×) with dense infiltration by a plasmablastic lymphoma. (B) Immunohistochemical analysis with CD20 positivity of 30%. (C, D) Association with EBV shown by positivity for EBER (C, Epstein–Barr encoded RNA) and EBNA (D, Epstein–Barr nuclear antigen). (E, F) Immunohistochemical staining for kappa- and lambda-light chains (400×) shows a kappa:lambda ratio of 1:9 demonstrating PTLD not to be completely monoclonal. (G) Bone marrow (May–Gruenewald–Giemsa, 400×) infiltrated by an early lesion type of PTLD with plasmocytic hyperplasia. (H) Intestinal mucosa (Giemsa, 100×) densly infiltrated by a plasmablastic lymphoma.
Patient 2 was a 70-year-old EBV-seropositive man with T-prolymphocytic leukemia in first complete remission after treatment with alemtuzumab. He underwent allogeneic HSCT from a matched unrelated donor after reduced intensity conditioning with fludarabine and total body irradiation (TBI) of 2 Gray. Eight months post-transplant while under combined immunosuppression with cyclosporine A (150 mg/day) and prednisone (15 mg/day), he presented with acute abdominal pain, diarrhea, and pancytopenia as well as reactivation of EBV to 5.500 GEq/ml. Computed tomography scan and diagnostic laparotomy showed rapidly progressive intestinal lymphadenopathy, liver mass, and thickening of the colon. Histological analysis revealed EBER- and CD20-positive diffuse large B-cell lymphoma with a proliferation fraction of 80% involving the intestinal lymph nodes, intestine, and liver. The bone marrow was faintly infiltrated. Massive post-operative abdominal bleeding and rapid multi-organ failure led to death 8 months after HSCT before specific therapeutic measures (rituximab) for the PTLD could be taken.
Patient 3 was a 29-year-old EBV-seropositive man with SAA nonresponsive to ATG and cyclosporine A. He underwent allogeneic HSCT with two cord blood units after conditioning with fludarabine, cyclophosphamide, and TBI of 2 Gray. EBV reactivation of 1.400 GEq/ml 4 months after HSCT while under combined immunosuppression with tacrolimus (1 mg/day) and prednisone (5 mg/day), was asymptomatic and was monitored in weekly intervals. Seven months post-transplant EBV replication increased to 12.400 GEq/ml, while he was symptomatic with bilateral retrobulbar neuritis and progressive visual impairment. Ocular symptoms were most probably EBV-associated as alternative causes were excluded and treatment with 4 cycles of rituximab (4 × 375 mg/m2/week) led to complete resolution of visual symptoms, while EBV replication became undetectable.
Patient 4 was a 30-year-old EBV-seropositive woman with a third relapse of stage IV Hodgkin lymphoma 3 years after autologous transplantation. She underwent allogeneic HSCT of a fully matched unrelated EBV-seropositive donor after conditioning with fludarabine, melphalan, and ATG. She presented with cervical, mediastinal, and abdominal lymphadenopathy and swollen tonsils along with EBV reactivation to 178.000 GEq/ml 1.5 months post-transplant. Immunosuppression was at cyclosporine A 150 mg/day. After 2 cycles of rituximab (2 × 375 mg/m2/week), EBV replication was undetectable and enlarged lymph nodes regressed continuously in size over several months. The patient died 32 months after HSCT due to refractory intestinal and hepatic GvHD without evidence of active PTLD.
Patient 5 was a 21-year-old EBV-seropositive woman with pure white cell aplasia non-responsive to granulocyte colony stimulating factor (G-CSF). A first allogeneic bone marrow transplant from a fully matched unrelated EBV-seropositive donor after conditioning with cyclophosphamide and ATG was rejected after 1 month. G-CSF was restarted along with alemtuzumab (10 mg/day for 10 days) and cyclosporine A (350 mg/day). Pronounced cervical and abdominal lymphadenopathy and EBV reactivation of 875 GEq/ml developed 4 months post-transplant. Histological analysis revealed an EBER- and CD20-positive pleomorphic DLBCL with proliferation activity of 50–80%. Cyclosporine A was stopped and 2 cycles of rituximab (2 × 375 mg/m2/week) were applied. The patient declined chemotherapy with R-CHOP and directly proceeded to a second allogeneic BMT of the same donor. PTLD clinically regressed and EBV replication became undetectable.
Discussion
We report the courses and outcome of PTLD related to EBV reactivation or EBV primary infection after allogeneic HSCT over a period of 4.5 years at our transplant center. Patients with symptomatic PTLD uniformly had unrelated stem cell donors, had received ATG and/or fludarabine for conditioning, and had all developed PTLD within 1 year post-transplant. Although these patients were similar in terms of stem cell donors and conditioning, the courses of PTLD in these individuals displayed a high diversity. Two of the five patients had a rapidly fatal course refractory to intense chemotherapy, while the three other patients showed a benign course with prompt response to anti-CD20 immunotherapy.
So far, severity of the course and outcome of PTLD cannot be anticipated. Promising therapies such as DLIs or EBV-specific CTLsCitation8–Citation10 require 2–3 weeks for production and delivery and thus often might not be available in time as in our patient 1. To take advantage of these cellular therapies, early risk stratification would be beneficial. Our data show that the level of EBV replication does not consistently associate with the severity of PTLD course. We find pronounced thrombocytopenia at the time of PTLD diagnosis in the patients with a detrimental course in contrast to borderline normal or normal platelet counts in the patients with a benign course of PTLD. A potential association of lymphopenia at the time of PTLD diagnosis with an adverse outcome is less evident, as three patients were lymphopenic, whereas two of them had a fatal course of PTLD. Primary EBV infection after transplantation might have added to the aggressive course in one patient. Therefore, seroconstellation for EBV between the stem cell donor and the recipient might also be critical in regards to PTLD disease severity. The relevance of these candidate factors needs to be studied in larger cohorts of patients with EBV-PTLD.
Treatment of PTLD has been importantly strengthened by anti-CD20 immunotherapy. Today, rituximab is an established and essential part in the treatment of PTLD after HSCT as well as after solid organ transplantation of the kidney, liver, and heart.Citation11–Citation14 In a recent study by Coppoletta et al.,Citation15 55 patients after allogeneic HSCT were treated with rituximab when EVB DNAemia was >1000 EBV copies/105 peripheral blood mononuclear cells. Overall, 50 of 55 patients (91%) cleared EBV after one dose (n = 25) or more than one dose (n = 25) of rituximab. The study showed the effectiveness of rituximab in controlling EBV DNAemia in patients undergoing allogeneic HSCT.
In addition, targeted cellular therapies such as EBV-specific CTLs are being developed. But still – also in the era of rituximab – PTLD is related to considerable mortality. Thus, patients at risk for PTLD require a very high level of clinical alertness with close monitoring of EBV viral load and extensive histological evaluation as soon as PTLD is suspected.
Our data demonstrate that the course of PTLD is highly diverse in patients at risk. Therefore, early detection of predictors for severe disease is crucial for planning treatment strategy. Based on our findings, we suggest further evaluation of thrombocytopenia, lymphopenia, and serological EBV constellation in regards to disease severity in EBV-PTLD.
References
- Curtis RE, Travis LB, Rowlings PA, Socié G, Kingma DW, Banks PM, et al. Risk of lymphoproliferative disorders after bone marrow transplantation: a multi-institutional study. Blood 1999;94:2208–16.
- Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343:481–92.
- van Esser JW, van der Holt B, Meijer E, Niesters HG, Trenschel R, Thijsen SF, et al. Epstein-Barr virus (EBV) reactivation is a frequent event after allogeneic stem cell transplantation (SCT) and quantitatively predicts EBV-lymphoproliferative disease following T-cell-depleted SCT. Blood 2001;98:972–8.
- Heslop HE. How I treat EBV lymphoproliferation. Blood 2009;114:4002–8.
- Glotz D, Chapman JR, Dharnidharka VR, Hanto DW, Castro MC, Hirsch HH, et al. The Seville expert workshop for progress in posttransplant lymphoproliferative disorders. Transplantation 2012;94:784–93.
- Styczynski J, Reusser P, Einsele H, de la Camara R, Cordonnier C, Ward KN, et al.; Second European Conference on Infections in Leukemia. Management of HSV, VZV and EBV infections in patients with hematological malignancies and after SCT: guidelines from the Second European Conference on Infections in Leukemia. Bone Marrow Transplant. 2009;43:757–70.
- Kamani NR, Kumar S, Hassebroek A, Eapen M, LeRademacher J, Casper J, et al. Malignancies after hematopoietic cell transplantation for primary immune deficiencies: a report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2011;17:1783–9.
- Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010;115:925–35.
- El-Bietar J, Bollard C. T-cell therapies for Epstein-Barr virus-associated lymphomas. Pediatr Hematol Oncol. 2011;28:627–39.
- Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012;119:2644–56.
- Kamdar KY, Rooney CM, Heslop HE. Posttransplant lymphoproliferative disease following liver transplantation. Curr Opin Organ Transplant. 2011;16:274–80.
- Caillard S, Lamy FX, Quelen C, Dantal J, Lebranchu Y, Lang P, et al.; French Transplant Centers. Epidemiology of posttransplant lymphoproliferative disorders in adult kidney and kidney pancreas recipients: report of the French registry and analysis of subgroups of lymphomas. Am J Transplant. 2012;12:682–93.
- Khedmat H, Taheri S. Heart allograft involvement by posttransplant lymphoproliferative disorders: report from the PTLD. Int Surv Exp Clin Transplant. 2011;9:258–64.
- Senechal M, Demers S, Cantin B, Bourgault C, Leblanc MH, Morin J, et al. Usefulness and limitations of rituximab in managing patients with lymphoproliferative disorder after heart transplantation. Exp Clin Transplant. 2012;10:513–8.
- Coppoletta S, Tedone E, Galano B, Soracco M, Raiola AM, Lamparelli T, et al. Rituximab treatment for Epstein-Barr virus DNAemia after alternative-donor hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2011;17:901–7.