
Abstract
Objectives
To determine whether the values of mean cell volume (MCV) and mean sphered cell volume (MSCV) can distinguish hereditary spherocytosis (HS) from thalassemia.
Methods
The MCV, MSCV, and other erythrocyte indexes were measured in totally 263 people, 57 HS patients, 109 thalassemia patients, and 107 normal control subjects. All indexes were derived from measurements obtained by the Beckman–Coulter LH 750 Hematology Analyzer.
Results
The MSCV was significantly lower in the HS group compared with the thalassemia group (P < 0.001), but the MCV was significantly higher in the HS group compared with the thalassemia group (P < 0.001). Among 57 patients with HS, the MCV was higher than the MSCV in 56 patients. The MCV was lower than the MSCV in one patient combined with β-thalassemia. In the control and thalassemia groups, the MCV was lower than the MSCV.
Conclusion
Measurements of the MCV higher than the MSCV can be considered an ideal index to distinguish rapidly HS from thalassemia.
Introduction
Hereditary spherocytosis (HS) is a hereditary disease, whose signs and symptoms include anaemia, jaundice, and splenomegaly.Citation1 The clinical severity of HS varies from asymptomatic carriers to severe haemolysis.Citation2 Approximately 20–30% of patients with mild HS have compensated haemolysis, and the production and destruction of erythrocytes were complemented or nearly equivalent.Citation3 These patients are not anaemic and are usually asymptomatic with mild splenomegaly, scant presence of spherocytes, and slightly affected reticulocytosis. Approximately 3–5% of severe HS patients suffer from severe anaemia and without regular transfusions or splenectomy the patients grow slowly, suffer from extramedullary haemopoiesis and bone changes – e.g. thalassaemia faces.Citation1 Thalassemia is one of the most prevalent genetic diseases in China, it occurs with a high frequency in Guangxi Autonomous Region, the carrier rates of α-thalassemia and β-thalassemia are 15.0 and 4.8%, respectively. Most of the carriers with thalassemia are in good health, occasionally, they are mildly anaemic. In severe cases, it results in severe anaemia, splenomegaly, and jaundice.Citation4 We found that some HS patients were clinically misdiagnosed as thalassemia.
Thalassemia is characterized by microcytosis and hypochromic anaemia, patients with thalassemia always have low mean cell volume (MCV). Microspherocytosis is the morphological hallmark of HS, which is caused by loss of membrane surface area, some HS patients also had MCV <80 fl.Citation5 Mean cell haemoglobin concentration (MCHC) and red cell distribution width (RDW) are used to differentially diagnose HS.Citation6,Citation7 However, the MCHC is not markedly increased in some HS patients under the conditions of high reticulocyte counts. Therefore, it is not easy to distinguish HS from thalassemia using the MCHC. It has been shown that the mean sphered cell volume (MSCV), which is the average of the total red blood cells (RBCs) in the reticulocyte analysis, is always lower than the MCV in HS. This artificial volume was useful in screening for HS from haemolytic anaemias.Citation8,Citation9 There are no reports using these red cell parameters together as indicators for distinguishing between HS and thalassemia. Our study examined the usefulness of MCV and MSCV in the first stage of patient assessment.
Methods
This study was approved by the Ethics Committees of the First Affiliated Hospital, Guangxi Medical University in China. All subjects gave their informed consent to participate in the study. Anaemia patients were collected from the Departments of Hematology and Pediatrics, First Affiliated Hospital, Guangxi Medical University, China, from November 2011 to March 2013. HS was diagnosed according to clinical history, spherocytes on peripheral blood smears, increase in reticulocyte, and abnormal osmotic fragility (OF) test. The HS group contained 57 patients belonging to 44 unrelated families was investigated, including 34 male and 23 female, with a mean age of 24.37 ± 14.94 years. Thalassemia was diagnosed by haemoglobin (Hb) electrophoresis and DNA analysis. The thalassemia group contained 109 patients (84 α-thalassemia and 25 β-thalassemia), including 38 male and 71 female, with a mean age of 25.39 ± 17.11 years. The normal control group contained 107 subjects, including 47 male and 60 female, with a mean age of 26.80 ± 10.59 years. Normal controls were healthy individuals age matched to the anaemia patients. Red cell parameters were detected using Beckman–Coulter LH 750 Hematology Analyzer (Beckman Coulter Inc, Fullerton, CA, USA). Red cell parameters contained RBC, Hb, haematocrit (Hct), MCV, MCH, MCHC, RDW, and MSCV.
Red cell parameters were analysed using Student's t-test, Kruskal–Wallis one-way analysis of variance post hoc tests, and Bonferroni test. A value of P < 0.05 was considered statistically significant.
Results
Statistical results of red cell parameters in the control, HS and thalassemia groups are shown in . The HS group showed significantly reduced (P < 0.001) RBC, Hb, Hct, MCV, and MSCV, but significantly increased (P < 0.001) RDW and MCHC compared with the control group. The thalassemia group had significantly lower (P < 0.001) Hb, Hct, MCV, MCH, MCHC, and MSCV, but significantly increased (P < 0.001) RDW compared with the control group. The HS group showed significantly increased (P < 0.001) MCV, MCH, and MCHC, but significantly lower (P < 0.001) RBC and MSCV compared with the thalassemia group.
Table 1. Summary statistics (mean ± SD) for the red cell indices for normal controls, and patient groups (HS and thalassemia)
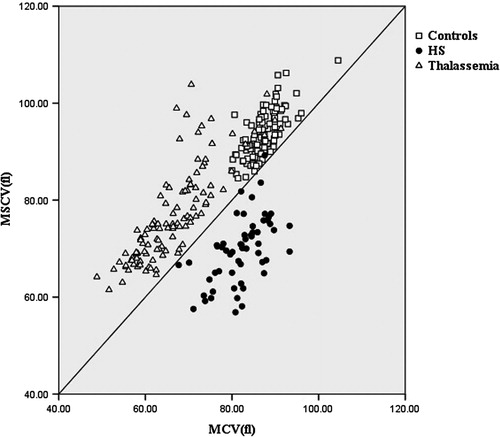
We set a standard curve according to MCV = MSCV. Data of the MCV and MSCV of all subjects are shown in . In the HS group, the MCV was higher than the MSCV in 56 patients. The MCV was lower than the MSCV in one patient combined with β-thalassemia. This 17-year-old female patient suffered from icteric sclera, which was aggravated when she had a cold, but with the absence of splenomegaly. Peripheral blood smears from the patient exhibited abundant microspherocytes and some lightly stained erythrocytes. Erythrocyte OF increased. Laboratory investigations revealed Hb 12.71 g/dl, RDW 15%, MCHC 31.78 mg/dl, MCH 25.62 pg/cell, MCV 73.55 fl, MSCV 75.23 fl, reticulocyte counts 2.0%. Hb electrophoresis showed Hb A2 5.6% and Hb F 0.8%. Genotype analysis of β-globin gene revealed that the patient was a heterozygote for codons 41–42 (-TCTT) mutation. In contrast, the MCV was lower than the MSCV in the control and thalassemia groups. The results show the comparison of MCV and MCV was significantly different between HS and thalassemia. The sensitivity of the ratio was 98.25% and specificity was 99.10% in separating HS from thalassemia.
Figure 1. Scatter plot for mean sphered cell volume (MSCV) vs. mean cell volume (MCV) for normal controls (n = 107) and patient groups, hereditary spherocytosis (HS) (n = 57) and thalassemia (n = 109).
Discussion
Conventional screening tests for HS mainly contain peripheral blood smear examination and the OF test. The OF test is time consuming, labour intensive, and adds nothing to the diagnosis if there are obvious spherocytes on the film. It can also be falsely negative in the presence of iron deficiency and obstructive jaundice.Citation10 Erythrocyte parameters such as MCHC and RDW have been used to screen HS.Citation6,Citation7 It was reported that the sensitivity and specificity of diagnosing HS were respectively 50 and 100% when the MCHC > 35.4 g/dl and the RDW > 14% in 112 HS child patients.Citation7 This study confirmed the MCHC increase was not significant in some HS patients under conditions of high reticulocyte counts. Excessive bilirubin levels in haemolytic anaemia patients interfered with Hb detection causing a false increase in MCHC.Citation11,Citation12 Thus, we attempted to identify other parameters to distinguish rapidly HS from thalassemia. Measurements of the MCV higher than the MSCV had been shown to be useful for the diagnosis of HS. Chiron et al.Citation8 reported that the MCV was higher than the MSCV had 100% sensitivity and 93.30% specificity for identifying the diagnosis of HS. In addition, Broseus et al.Citation9 observed that the delta (MCV–MSCV) values >9.6 fl showed 100% sensitivity and 90.57% specificity in discriminating HS from other subjects.In comparison, in our results, 56 (98.25%) of 57 HS patients had the MCV higher than the MSCV and the delta (MCV–MSCV) was >9.6 fl in 44 (77.19%). In patients without HS, the MCV was higher than the MSCV was not rare. The case that MCV was higher than the MSCV can also presented by other kinds of anaemia, such as autoimmune haemolytic anaemia, but it can be rule out by the coombs test and flow cytometric test.
This study retrospectively analysed the parameters of erythrocytes and reticulocytes in patients with HS and thalassemia. Results demonstrated that only 29 patients in the HS group had MCHC >35.4 g/dl and RDW >14%. Four subjects in the thalassemia group and two in the control group achieved these requirements. One patient from the HS group combined with thalassemia had the MCV lower than the MSCV. HS combined with β-thalassemia is a rare occurrence. Erythrocyte surface-to-volume ratio increased in thalassemia patients, but erythrocyte surface-to-volume ratio decreased and Hb content increased in HS patients. Thus, one abnormality was compensated by another abnormality.Citation13 A previous study showed that this mechanism only changed the curve of erythrocyte OF, but did not affect the clinical manifestations.Citation14 Results from this study confirmed that the comparison of MCV and MSCV is highly effective in screening haemolytic anaemia, confirming a previous study.Citation8,Citation9
In summary, for clinicians treating haemolytic anaemia patients, the MCV and MSCV can be used to rapidly distinguish HS from thalassemia. If the MCV is higher than the MSCV, it can be considered as HS.
Acknowledgement
This study was supported by the National Natural Science Foundation of China (No. 81360263).
References
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet 2008;372:1411–26.
- Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. Guidelines for the diagnosis and management of hereditary spherocytosis–2011 update. Br J Haematol. 2012;156:37–49.
- Guarnone R, Centenara E, Zappa M, Zanella A, Barosi G. Erythropoietin production and erythropoiesis in compensated and anaemic states of hereditary spherocytosis. Br J Haematol. 1996;92:150–4.
- Pan HF, Long GF, Li Q, Feng YN, Lei ZY, Wei HW, et al. Current status of thalassemia in minority populations in Guangxi, China. Clin Genet. 2007;71:419–26.
- Bunyaratvej A, Bunyaratvej P. Measurements of cell volume and hemoglobin concentration of erythrocytes from hereditary ovalocytosis and hereditary spherocytosis. J Med Assoc Thai. 1992;75( Suppl 1):248–52.
- Christensen RD, Henry E. Hereditary spherocytosis in neonates with hyperbilirubinemia. Pediatrics 2010;125:120–5.
- Michaels LA, Cohen AR, Zhao H, Raphael RI, Manno CS. Screening for hereditary spherocytosis by use of automated erythrocyte indexes. J Pediatr. 1997;130:957–60.
- Chiron M, Cynober T, Mielot F, Tchernia G, Croisille L. The GEN.S: a fortuitous finding of a routine screening test for hereditary spherocytosis. Hematol Cell Ther. 1999;41:113–6.
- Broseus J, Visomblain B, Guy J, Maynadie M, Girodon F. Evaluation of mean sphered corpuscular volume for predicting hereditary spherocytosis. Int J Lab Hematol. 2010;32:519–23.
- Bolton-Maggs PH. Hereditary spherocytosis; new guidelines. Arch Dis Child. 2004;89:809–12.
- Kar R, Sharma CB. Bilirubin peak can be mistaken as Hb Bart's or Hb H on High-performance liquid chromatography. Hemoglobin 2011;35:171–4.
- Wong SS, Schenkel OJ. Quantification of plasma hemoglobin in the presence of bilirubin with bilirubin oxidase. Ann Clin Lab Sci. 1995;25:247–51.
- Miraglia del Giudice E, Perrotta S, Nobili B, Pinto L, Cutillo L, Iolascon A. Coexistence of hereditary spherocytosis (HS) due to band 3 deficiency and beta-thalassaemia trait: partial correction of HS phenotype. Br J Haematol. 1993;85:553–7.
- Pautard B, Feo C, Dhermy D, Wajcman H, Baudin-Chich V, Delobel J. Occurrence of hereditary spherocytosis and beta thalassaemia in the same family: globin chain synthesis and visco diffractometric studies. Br J Haematol. 1988;70:239–45.