
Abstract
Introduction
Fetal hemoglobin (HbF) is the predominant hemoglobin in red cells during fetal life. Just after birth, the level of HbF decreases gradually to <1%, and is replaced mainly by adult hemoglobin (HbA) (∼97%). However, higher HbF levels could be associated with HbE/β-thalassemia, a complex thalassemia intermedia with a diverse clinical severity ranging from mild-to-severe anemia. This study investigates the correlation of HbF level with the clinical and laboratory data of HbE/β-thalassemia individuals.
Methods
Peripheral blood samples from 30 HbE/β-thalassemia subjects were subjected to a full blood count, genomic as well as quantitative real-time polymerase chain reaction gene expression studies. Statistical analyses were performed using SPSS 17.0.
Results
HbF levels were influenced by age, mean cell volume (MCV), mean cell hemoglobin (MCH), HbA, β-globin, and α/β-globin expressions.
Discussion
HbF production is affected by the α/β-globin chain imbalance due to the lack of β-globin gene expression as well as inversely correlates to the amount of functional hemoglobin available in the cells.
Introduction
During fetal life, fetal hemoglobin (HbF, α2γ2) is the main hemoglobin subtype to carry oxygen before it is replaced by adult hemoglobin (HbA, α2β2) as the predominant hemoglobin, which occurs just after birth. This hemoglobin switching is attributed by the replacement of γ-globin chains to β-globin chains while α-globin chains remain the same. Thereafter, a minute presence of HbF (<1%) remains and HbA constitutes ∼97% of the hemoglobin in the adult blood circulation.Citation1 So any defects in the β-globin gene become clinically apparent as HbF is slowly substituted by HbA.
In Malaysia, HbE/β-thalassemia is the most prevalent β-thalassemia intermedia in the Malay ethnicity.Citation2 There is a diverse clinical severity as a consequence of gene modifiers, environmental factors, and lifestyle. There are three categories of genetic modifiers that play a role in influencing the clinical manifestations, i.e. primary modifiers (specific β-alleles that affect the degree of β-globin chains deficiency); secondary modifiers (affecting the α/β-globin chain imbalance); and tertiary modifiers (affecting the disease progress).Citation2,Citation3 The severity of β-thalassemia may be alleviated by HbF production as it can alter the α/β-globin chain imbalance.Citation3,Citation4 Therefore, HbF has been studied extensively from genomic to clinical applications in order to modulate the pathophysiology of β-thalassemia.
Our aim here is to convey an understanding of HbF by correlating it with demographic data, clinical data, genotypes, and globin gene expressions in HbE/β-thalassemia individuals.
Materials and methods
Study subjects
Thirty recorded HbE/β-thalassemia individuals from Hospital Ampang Thalassemia Clinic and University Malaya Medical Centre were recruited for this study. Subjects with α-thalassemia, iron deficiency anemia, hydroxyurea therapy, or transfused <3 months prior to our study were excluded to reduce confounding factors that may affect the results. This study was approved by the Medical Research and Ethics Committee, Ministry of Health Malaysia (KMM/NIHSEC/08/0804/P09-341), Medical Ethics Committee, University Malaya Medical Centre (739.4) and Medical Research Ethics Committee, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia (UPM/FPSK/PADS/T7-MJKEtikaPer/F01(LECT_JUN(08)10). Informed consent was given prior to blood collection and all data were anonymized with numerical identification throughout the study.
Blood analysis
Complete blood count analysis and high performance liquid chromatography (HPLC) were performed on collected peripheral blood samples using Sysmex® XE-5000 (Sysmex®, Kobe, Japan) and VARIANT II β-Thalassemia Short Program on VARIANT II™ Hemoglobin Testing System (Bio-Rad Laboratories, Hercules, CA, USA), respectively. Plasma ferritin was used for iron deficiency anemia screening (Tina-quant® Ferritin assay, Cobas, Roche Diagnostics GmbH, Mannheim, Germany).
Genotype studies
Common Malaysian β-thalassemia mutations including −29 (A > G), −28 (A > G), codon 8/9 (+G), codon 17 (A > T), codon 19 (A > G), IVS I-5 (G > C), IVS I-1 (G > T), codon 41/42 (-TCTT), and IVS II 654 (C > T) were genotyped by amplification refractory mutation system polymerase chain reaction (ARMSPCR) using selected primer sequences and PCR protocol modified from Old et al.Citation5 Briefly, total reaction volume contains 1X standard Taq reaction buffer, 1.5 mM MgCl2, 200 µmol/l dNTPs, 0.5 U Taq DNA polymerase (BioLabs® Inc., New England, USA), and 150 ng genomic DNA. β-Actin was used as an internal control with the sequences taken from Bergmann et al.Citation6 The cycling condition was an initial 5 minutes denaturation step at 94°C followed by 30 cycles of 94°C for 1 minute, annealing temperature for 1 minute, and 72°C for 1.5 minutes extension step. The cycling condition ends with a final 72°C extension step for 5 minutes. Any uncharacterized samples were sent for sequencing.
Co-inheritance of α-thalassemia was excluded by /--SEA and /-α3.7 mutation screening using standard multiplex PCR protocol by Liu et al.Citation7 The multiplex involves 1X GeneAmp® PCR gold buffer, 200 µmol/l GeneAmp® dNTP mix, 1.5 mmol/l MgCl2 solution, 1.25 U AmpliTaq® Gold DNA polymerase (Applied Biosystems, Roche Molecular Systems Inc., Branchburg, NJ, USA), 0.75 mol/l betaine solution, 5% dimethyl sulfoxide (Sigma®, Taufkirchen, Germany), and 10 ng genomic DNA. Xmn1 polymorphism was characterized through restriction fragment length polymorphism assay as described in Wong et al. (2006).Citation8 A standard PCR protocol was done followed by Xmn1 digestion of the amplified DNA according to manufacturer's protocol (FastDigest® Pdm1 (Xmn1), Fermentas Inc., Hanover, Maryland, USA).
Expression studies
RNA extracted from enriched peripheral reticulocytes as described in Lai et al.Citation9 were reverse transcribed using QuantiTect® Reverse Transcription Kit according to manufacturer's protocol (QIAGEN® GmbH, Hilden, Germany). To enrich the reticulocytes, the peripheral blood sample was centrifuged to separate into three distinct layers of plasma, mononuclear cells, and erythrocytes. The plasma and mononuclear cell layers were removed and the erythrocytes were washed three times with phosphate-buffered saline (Sigma®). A final spin of 30 minutes redistributed the reticulocytes to the top for leucodepletion process and RNA extraction. Expressions of α-, β-, and γ-globin genes were analyzed with GAPDH as the housekeeping gene using TaqMan® assays (PrimeTime™ Mini qPCR Assay, IDT®, Singapore, Singapore and Applied Biosystems, Warrington, UK).
Statistical analysis
Student's t-test, analysis of variance, simple linear regression, and Pearson's correlation coefficient analysis were performed using SPSS 17.0 for mean comparison and correlation studies.
Results
HbF level and demographic data
The 30 HbE/β-thalassemia samples comprised of 12 males and 18 females of which 26 were of Malay ethnicity and 4 Chinese, ranging from 15 to 51 years old were collected. There was no evidence of α-thalassemia or iron deficiency anemia in the samples. HbF level ranges from 5.9 to 49.1% with a positive correlation to age (P = 0.020) while no association was found between HbF and gender (P = 0.205).
Analysis of HbF level with hematological data and genotypes
Simple linear regression analysis on red blood cell (RBC) indices showed no significant associations between HbF and RBC count (P = 0.086), hemoglobin level (P = 0.563), hematocrit (P = 0.665), mean corpuscular hemoglobin concentration (MCHC) (P = 0.728), red cell distribution width (P = 0.633), and reticulocytes count (P = 0.617). RBC indices that were positively correlated to HbF are mean corpuscular volume (MCV) (P = 0.008) and MCHC (P = 0.013).
HbF (%) was significantly associated with HbA (%) level (P = 0.002) and absolute HbF (g/dl) (percentage HbF of total hemoglobin (g/dl)) was also significantly correlated to absolute HbA level (g/dl) (P = 0.017). Xmn1 and β-thalassemia genotypes were not associated to HbF level.
Analysis of HbF level with globin gene expressions and splenectomy history
HbF was further correlated to α-, β-, and γ- globin gene expressions including their derivatives using regression analysis. All expression data were positively skewed, thus had to be log-transformed to fit the normal distribution. HbF was significantly associated with α/β expression ratio (P = 0.005) with a near-significant association with β-globin gene expression (P = 0.059). There were no correlation of HbF to α-globin (P = 0.328), γ-globin (P = 0.077), and excess α-globin (P = 0.276) expressions. HbF was also not correlated to splenectomy history (P = 0.920).
All data are summarized in Tables and and .
Figure 1. Clinical and laboratory data significantly correlated to HbF level. (A) HbF and age (P = 0.020); (B) HbF and MCV (fl) (P = 0.008); (C) HbF and MCH (pg) (P = 0.013); (D) absolute HbF and absolute HbA (g/dl) (P = 0.017); (E) HbF with a near-significant correlation to log β-globin expression (P = 0.059); and (F) HbF and log α/β-globin expression (P = 0.005).
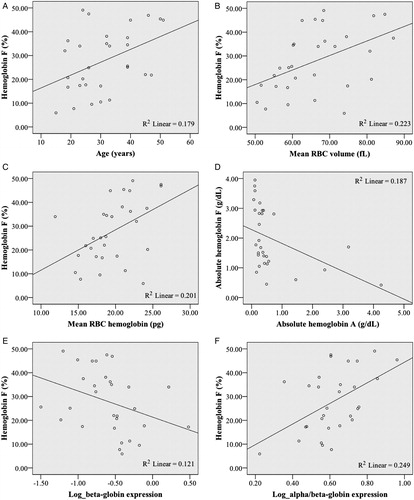
Table 1. Mean comparison of HbF level (%) in relation to demographic data, splenectomy history, and genotypes
Table 2. Correlation of HbF level (%) with age, hematological data, and globin gene expressions and their derivatives
Discussion
β-Thalassemia patients have defective β-globin production leading to an α/β-globin imbalance. The relative excess of α-globins will exert erythropoietic stress onto the red cell membrane through precipitation and reactive oxygen species generation.Citation10 In β-thalassemia patients, elevated γ-globin expression will reduce the α/β-globin chain imbalance by increasing the HbF synthesis. This would lead to a milder clinical presentation in β-thalassemia intermedia, despite similar beta genotypes and normal alpha genotypes.Citation3
Our HbE/β-thalassemia cohort displayed a considerable range of HbF level, from 5.9 to 49.1%. This wide range of HbF has been reported by many groups albeit with variable mean levels.Citation11–Citation13 This high variability could be due to the subjective HbF production which has been shown to be largely genetically controlled.Citation14
To exclude the possible biasness of delayed hemoglobin switching, the subjects were patients between the ages of 15 and 51 years. Age upon blood collection showed a significant positive correlation of HbF level in our study. We believe that selective increased production of HbF in some individuals gives them an added survival advantage as even a modest elevation of HbF level gives a therapeutic value to HbE/β-thalassemia patients.Citation12 Another study has shown that HbF levels have a negative correlation to age which they have not been able to explain, but this difference could be attributed to the fact that they have a younger cohort of patients compared to ours.Citation11 Studies have identified three quantitative trait loci, Xmn1-HBG2, HBS1 L-MYB intergenic polymorphism locus, and BCL11A, as major contributors of HbF variations (20–50%) in sickle cell anemia and healthy individuals of European descent.Citation4 Some studies have shown a linkage between Xmn1 polymorphisms and certain β-globin haplotypes and genotypes although similar to us, other groups demonstrated no association.Citation7,Citation13,Citation15–Citation16 The levels of HbF could be controlled by other genetic factors, for example, by those found in BCL11A and HBS1 L-MYB genes.Citation17–Citation19 Effects of these genes in the Malaysian population have not been studied. Perhaps, the higher HbF levels in some patients compared to others could be contributed by these loci leading to a less severe phenotype and prolonged lifespan.
Musallam et al.Citation20 has reported splenectomized patients have significantly higher HbF levels compared to non-splenectomized (P = 0.007) and an improvement in growth and development was seen in splenectomized individuals in a study by Olivieri et al. (2008).Citation21 We hypothesized an increase in HbF level post-splenectomy due to less splenic consumption and more F cells available for functional purposes. However, such association was not observed.
Both mean red cell size (MCV) and mean hemoglobin content per red cell (MCH) were positively correlated with HbF level suggesting that increased HbF level improved the erythropoietic environment significantly in HbE/β-thalassemia. Sharma et al.Citation22 and Kishore et al.Citation23 reported negative correlations between HbF level with MCV and MCH, respectively, but their study subjects included various groups of HbE diseases.
An inverse correlation was found to exist between HbF and HbA levels, similar to a study by Dover and Boyer,Citation24 which demonstrated a reciprocal relationship between HbA and HbF production in adults. Our result is in agreement with Karlsson and NienhuisCitation25 that even a small augmentation of absolute HbF will result in cells that have less severe chain imbalance and thus have a preferential production and survival advantage in the bone marrow as well as in the circulation.
There was a near-signficant negative correlation of HbF to the β-globin expression and a significant correlation between HbF and log α/β-globin expression but not to log excess α-globin nor γ-globin expression. These suggest that HbF corresponds to the lack of β-globin expression rather than excess α-globin expression or higher expression of γ-globin gene.
In conclusion, our study suggests that HbF is a compensatory mechanism that responds directly to the level of HbA and β-globin gene expression. However, the genetic factors that modulate this compensation mechanism has yet to be elucidated in the Malaysian population.
Acknowledgements
The authors wish to thank the Director General of Health, Malaysia for permission to publish this paper.
Disclaimer statements
Contributors WFL collected the samples, performed the experiments, and wrote the paper, LM collected the data, EG provided expert consultation, JS contributed the samples and expert consultation, LKT provided technical expertise, and MIL designed the experiments and wrote the paper.
Funding This work was supported by the Fundamental Research Grant Scheme, Ministry of Higher Education, Malaysia (03-1-07-324FR and 04-04-10-837FR) awarded to M.I.L.
Conflicts of interest None.
Ethics approval This study was approved by the Medical Research and Ethics Committee, Ministry of Health Malaysia (KMM/NIHSEC/08/0804/P09-341), Medical Ethics Committee, University Malaya Medical Centre (739.4) and Medical Research Ethics Committee, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia (UPM/FPSK/PADS/T7-MJKEtikaPer/F01(LECT_JUN(08)10).
References
- Wood WG. Increased HbF in adult life. Bailliere Clin Haematol. 1993;6:177–213.
- George E. HbE/β-thalassemia in Malaysia: revisited. J Hematol Thromboembolic Dis. 2013;1:101. doi:10.4172/2329-8790.1000101
- Thein SL. Genetic modifiers of the β-hemoglobinopathies. Br J Haematol. 2008;141:357–66.
- Thein SL, Menzel S. Discovering the genetics underlying foetal hemoglobin production in adults. Br J Haematol. 2009;141:357–66.
- Old JM, Varawalla NY, Weatherall DJ. Rapid detection and prenatal diagnosis of beta-thalassaemia: studies in Indian and Cypriot populations in the UK. Lancet. 1990;336:834–7.
- Bergmann L, Miething C, Maurer U, Brieger J, Karakas T, Weidmann E, et al. High levels of Wilms’ tumor gene (wt1) mRNA in acute myeloid leukemias are associated with a worse long-term outcome. Blood 1997;90:1217–25.
- Liu RR, Wang MY, Lai YR. Analysis of Gγ-158(C > T) polymorphism in hemoglobin E/β-thalassemia major in Southern China. J Hematol Oncol. 2010;3:29.
- Wong YC, George E, Tan KL, Yap SF, Chan LL, Tan JAMA. Molecular characterization and frequency Gγ Xmn I polymorphism in Chinese and Malay β-thalassaemia patients in Malaysia. Malaysian Journal of Pathology. 2006;28:17–21.
- Lai MI, Jiang J, Silver N, Best S, Menzel S, Mijovic A, et al. Alpha-haemoglobin stabilising protein is a quantitative trait gene that modifies the phenotype of beta-thalassaemia. Br J Haematol. 2006;133:675–82.
- Thein SL. Genetic insights into the clinical diversity of beta thalassaemia. Br J Haematol. 2004;124:264–74.
- Rees DC. Hemoglobin F and hemoglobin E/β-thalassemia. J Pediatr Hematol Oncol. 2000;22:567–72.
- Allen A, Fisher C, Premawardhena A, Peto T, Allen S, Arambepola M, et al. Adaptation to anemia in hemoglobin E-β thalassemia. Blood 2010;116:5368–70.
- Nuntakarn L, Fucharoen S, Fucharoen G, Sanchaisuriya K, Jetsrisuparb A, Wiangnon S. Molecular, hematological and clinical aspects of thalassemia major and thalassemia intermedia associated with Hb E/β-thalassemia in Northeast Thailand. Blood Cells Mol Dis. 2009;42:32–5.
- Garner C, Tatu T, Reittie JE, Littlewood T, Darley J, Cervino S, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood 2000;95(1):342–6.
- Mosca A, Paleari R, Leone D, Ivaldi G. The relevance of hemoglobin F measurement in the diagnosis of thalassemias and related hemoglobinopathies. Clin Biochem. 2009;42:1797–801.
- Neishabury M, Azarkeivan A, Najmabadi H. Frequency of positive XmnI Gγ polymorphism and coinheritance of common alpha thalassemia mutations do not show statistically significantly difference between thalassemia major and intermedia cases with homozygous IVSII-1 mutation. Blood Cells Mol Dis. 2010;44:95–9.
- Galanello R, Sanna S, Perseu L, Sollaino MC, Satta S, Lai ME, et al. Amelioration of Sardinian beta-zero thalassemia by genetic modifiers. Blood 2009;114:3935–7.
- Thein SL, Menzel S, Peng X, Best S, Jiang J, Close J, et al. Intergenic variants of HBS1 L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci USA. 2007;104:11346–51.
- Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA. 2008;105:1620–5.
- Musallam KM, Sankaran VG, Cappellini MD, Duca L, Nathan DG, Taher AT. Fetal hemoglobin levels and morbidity in untransfused patients with β-thalassemia intermedia. Blood. 2012;119(2):364–47.
- Olivieri NF, Muraca GM, O'Donnell A, Premawardhena A, Fisher C, Weatherall DJ. Studies in hemoglobin E beta-thalassaemia. Br J Hematol. 2008;141:388–97.
- Sharma A, Marwah S, Buxi G, Yadav R. Hemoglobin E syndromes: emerging diagnostic challenge in North India. Indian J Hematol Blood Transfus. 2012;29:21–5.
- Kishore B, Khare P, Gupta RJ, Bisht S, Majumdar K. Hemoglobin E disease in North Indian population: a report of 11 cases. Hematology. 2007;12:343–7.
- Dover GJ, Boyer SH. Quantitation of hemoglobins within individual red cells: asynchronous biosynthesis of fetal and adult hemoglobin during erythroid maturation in normal subjects. Blood. 1980;56:1082–91.
- Karlsson S, Nienhuis AW. Developmental regulation of human globin genes. Annu Rev Biochem. 1985;54:1071–108.