Abstract
The quorum sensor and signalling molecule pyocyanin (PYO) contributes significantly to the pathophysiology of Pseudomonas aeruginosa infections. Comparison to phenothiazine drugs suggests that the antimalarial compound methylene blue (MB) can be regarded as a sulfur analog of PYO. This working hypothesis would explain why the synthetic drug MB behaves as a compound shaped in biological evolution. Here we report on redox-associated biological and biochemical properties of PYO in direct comparison to its synthetic analog MB. We quantitatively describe the reactivity of both compounds toward cellular reductants, the reactivity of their reduced leuco-forms towards O2, and their interactions with FAD-containing disulfide reductases. Furthermore, the interaction of PYO with human glutathione reductase was studied in structural detail by x-ray crystallography, showing that a single PYO molecule binds to the intersubunit cavity of the enzyme. Like MB, also PYO was also found to be active against blood schizonts of the malaria parasite P. falciparum in vitro. Furthermore, both compounds were active against the disease transmitting gametocyte forms of the parasites, which was systematically studied in vitro. As shown for mice, PYO is too toxic to be used as a drug. It may, however, have antimalarial activity in numerous human patients with concomitant Pseudomonas infections. MB, in contrast to PYO, is well tolerated and represents a promising agent for MB-based combination therapies against malaria. Current and future clinical studies can be guided by the comparisons between MB and PYO reported here. Additionally, it is of interest to study if and to what extent the protection from malaria in patients with cystic fibrosis or with severe wound infections is based on PYO produced by Pseudomonas species.
Introduction
Phenazines, nitrogen-containing heterocyclic compounds synthesized by Pseudomonas species and a number of other bacteria, are secondary metabolites with important biological activities. Pyocyanin (5-methyl-1-hydroxyphenazine, PYO, A) is a blue, redox-active phenazine produced and secreted by Pseudomonas aeruginosa where it serves as a respiratory factor, a quorum sensor, and an anti-microbial molecule.Citation1–Citation4 P. aeruginosa as a leading bacterial pathogen in hospital settings is the primary cause of sepsis and death in patients immunocompromised as a result of neutropenia, burns, or cystic fibrosis. Concentrations of up to 100 µM PYO have been detected in the sputa of P. aeruginosa-infected cystic fibrosis patients.Citation5
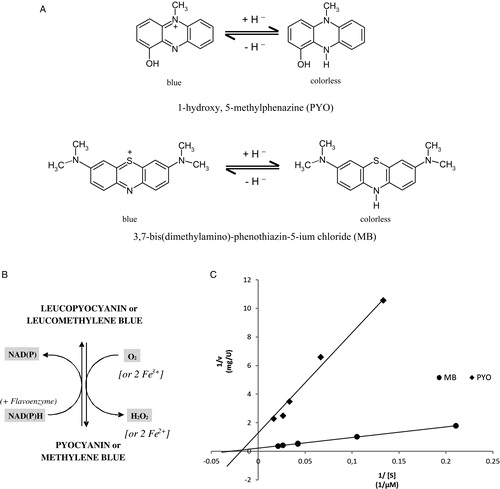
Figure 1. (A) Structures of PYO and MB in oxidized and reduced forms. (B) PYO and MB as redox cycling catalysts in vivo: NAD(P)H-dependent reduction of the blue oxidized forms and rapid re-oxidation of the leuco forms by auto-oxidation. (C) PYO and MB as diaphorase substrates of P. falciparum mitochondrial lipoamide dehydrogenase. In the Lineweaver–Burk diagram, the ordinate axis intercepts represent the reciprocal values of the specific activity for PYO (squares) and MB (circles). The enzyme preparation described in the experimental section was used. The OH group of PYOox has a pKa of 4.9.
PYO is known to exhibit pro-oxidant activity and, as a zwitterion with both hydrophobic and hydrophilic regions; it can easily interact with and penetrate cell-biological membranes at pH 7.0. PYO can directly accept electrons from reducing agents such as NADH, NADPH, and reduced glutathione (GSH). Electrons are then transferred to oxygen in order to generate reactive oxygen species (ROS) such as hydrogen peroxide and singlet oxygenCitation6 at the expense of host antioxidant capacity.Citation3 GSH levels of several cell lines exposed to PYO have been shown to be depleted by up to 50%Citation6,Citation7 and H2O2 production has been demonstrated to increase dramatically.Citation8 The concentration-, time-, and O2-dependent reactions of GSH with PYO were recently analyzed in detail by Cheluvappa et al.Citation9
Studies on the role of PYO in P. aeruginosa virulence in vivo using alternative model hosts revealed that PYO participates in the rapid killing of the nematode Caenorhabditis elegans by producing ROSCitation10 and severely affects numerous other organisms including bacteria,Citation11 yeast, fungi,Citation2 insects,Citation12 plants,Citation13 and mice.Citation3,Citation14 P. aeruginosa mutants lacking PYO are greatly attenuated in both acute and chronic infection as shown in mouse models.Citation3
In order to find physiological targets of PYO, Ran et al.Citation8 screened a Saccharomyces cerevisiae deletion library. Sixty percent of the hits have orthologs in humans and indicate that cell cycle, electron transport and respiration, epidermal cell growth, protein sorting, vesicle transport, and the vacuolar ATPase may be targeted. Beside these properties, which are mainly attributed to increased ROS levels, PYO shows physical effects affecting ciliary function and immunological effects interfering with interleukins, neutrophil apoptosis, and T cell response, for example. These effects might result in part from increased ROS levels (for a review see Liu and NizetCitation15 and references therein). However, the interaction of PYO with central cellular redox enzymes, including disulfide reductases like glutathione reductase (GR) and thioredoxin reductase (TrxR), has not yet been studied in sufficient detail.
The phenothiazine compound methylene blue (MB; methylthionine hydrochloride or 3,7-bis(dimethylamino)phenothiazin-5-ium chloride, A), the first synthetic compound to be used as a drug, was described by Guttmann and Ehrlich in 1891 as effective against malaria.Citation16 MB is registered in most countries as the treatment of choice for acute and chronic methemoglobinemia. It has been intensely studied and its numerous favorable properties have been described, including the differential staining of cell biological structures and protein crystals, its medicinal applications, and its unique physicochemical and photochemical characteristics.Citation17–Citation21 Like PYO, MB exhibits a remarkable reactivity toward cellular reductants and interacts with signaling proteins, flavoenzymes, haem proteins, or transmembrane transporters.Citation19,Citation20 Thus, MB behaves almost as a molecule shaped in biological evolution and indeed it has striking structural and functional similarities with PYO ().
Here we report – in the context of intracellular redox reactions – on further important properties of PYO and MB. This includes comparative studies on their redox cycling behavior, the quantitative reduction of PYO and MB by diaphorases, and the inhibition of disulfide reductases by PYO and MB. In addition to these kinetic data, we provide the x-ray structure of PYO bound to human GR. Furthermore, we show that both compounds are active against the human malaria parasite Plasmodium falciparum, including stage-specific effects and a remarkable activity against the disease-transmitting gametocytic forms.
Results
Redox cycling of PYO in the presence of intracellular reductants and triplet oxygen
Many pharmacological effects of PYO and MB are due to spontaneous or enzyme-catalyzed redox-cycling reactions (see Tables and and Citation19). The reducing equivalents required for these reactions are supplied by major intracellular reductants such as NADPH, NADH, or glutathione; the final electron receptor is often O2 (B). The rate constant for the spontaneous second-order reaction PYO + NADPH + H+ → LeucoPYO + NADP+ () was determined as previously described for MB and found to be 80 M−1 second−1 at pH 7.3 and 37°C; values for 25°C are given in ; assuming cytosolic concentrations of 40 µM NADPH and 25 µM PYO, the oxidation of NADPH would correspond to approx. 5 µM/minute. In order to slow down the spontaneous reaction between PYO and NAD(P)H in pharmacological experiments we recommend choosing a pH of 8.0.
Figure 2. Spectra of PYO in oxidized and NADPH-reduced form in 50 mM potassium phosphate buffer of pH 7. The peak at 340 nm in the reduced spectrum (dotted line) is largely due to excess NADPH.
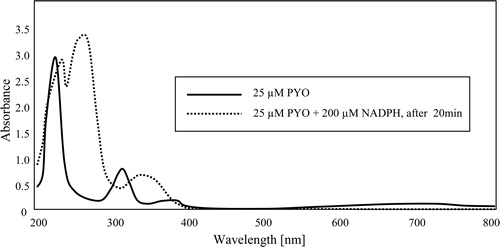
Table 1. Properties of PYO in comparison to MB
Table 2. Enzyme-kinetic data for interactions of PYO and MB with disulfide reductases
PYO reacts much more slowly than MB with intracellular thiol compounds such as thioredoxin or glutathione, both in the absence and presence of daylight.Citation19
Enzymatic reduction of PYO by diaphorases
In , PYO and MB are compared as diaphorase substrates (B and C) of disulfide reductases. Diaphorases or NAD(P)H:acceptor oxidoreductases [EC 1.6.5.2.] are enzymes that can catalyze the transfer of electrons from NAD(P)H to reducible aromatic compounds. A typical flavoprotein with strong diaphorase activity is dihydrolipoamide dehydrogenase (LipDH).Citation21 Both PYO and MB are reduced enzymatically and subsequently reoxidized in spontaneous reactions with oxygen. Each redox cycle results in the loss of NADPH and the production of toxic H2O2 (B). This pro-oxidant process contrasts with the physiological functions of the disulfide reductases which normally produce thiol groups as antioxidants. The flavoenzyme GR for instance catalyzes the reaction NADPH + GSSG + H+ → NADP+ + 2 GSH. GSSG and GSH stand for glutathione disulfide and reduced glutathione, respectively. TrxRs (NADPH + TrxS2 + H+ → NADP+ + Trx(SH)2) produce reduced thioredoxin, and the back reaction of LipDH (NADH + lipoamide + H+ → NAD+ + dihydrolipoamide) can contribute reducing equivalents in the form of dihydrolipoamide. These thiol-producing systems work in parallel but, via redoxins, they can closely interact and support each other in a number of pathophysiological situations.28
PYO as an inhibitor of disulfide reductases
Like MB, PYO can interact with enzymes not only as a substrate but also as a dissociable inhibitor (). The binding site for PYO as an inhibitor of human GR, shown in and , is described in detail below. In contrast to MB, PYO is a better inhibitor of human erythrocytic GR (see x-ray structure) than e.g. of P. falciparum GR (see IC50 values in ). The IC50 values referring to the inhibition of the GR-catalyzed reaction can be interpreted as Ki values if the inhibition type is non-competitive.Citation27,Citation28 This means that the inhibitor binds only to the free enzyme, not to enzyme–substrate complexes. While the enzyme kinetics clearly indicated non-competitive inhibition under standard conditions, a change from pH 7.3 to pH 8.0 led to an uncompetitive inhibition type as shown by Cornish-Bowden and Dixon-diagramsCitation29 (data not shown). In these studies, both the concentration of the substrate GSSG (in the range between 58 and 390 µM) and the PYO concentration (between 0 and 10 µM) were varied. The Ki’ determined for this uncompetitive inhibition was 7.5 µM for human GR. This means that at pH 8.0 the inhibitor PYO does not bind to the free enzyme but rather to the enzyme–substrate complex ES. Like MB, PYO does not inhibit the LipDHs of P. falciparum and man ().
Figure 3. Human GR homodimer with bound PYO. PYO (blue) and FAD (yellow) are represented as ball-and-stick models. Additionally, the surfaces of the catalytic cysteines Cys58/Cys63 and Cys58′/Cys63′ (yellow) and of PYO (blue) are shown. The atomic coordinates and their structure factors determined by x-ray diffraction analysis have been deposited at the PDB (Protein Data Bank) under the accession code (PBD ID) 3SQP. Molecular graphics images were produced using the UCSF Chimera package.Citation26
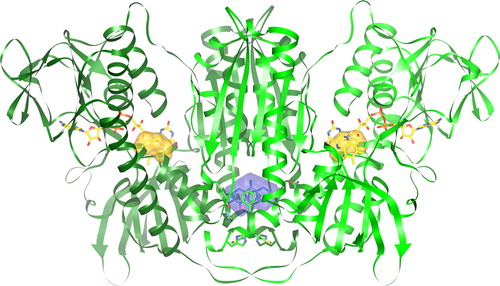
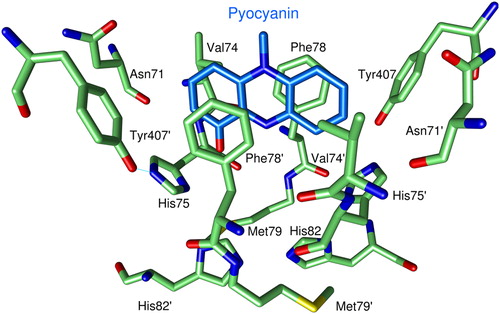
Figure 4. Binding of PYO to human GR. For clarity only one of the two observed orientations of PYO (blue carbon atoms) is shown. Green Cα-atoms indicate amino acids at distances of less than 5 Å from the ligand. O atoms are shown in red, N atoms in blue, and sulfur atoms in yellow.
The GR inhibition observed in vitro can be of pathological significance since rather high (up to 100 µM) extracellular PYO concentrations have been reported for infections caused by the PYO-producing bacterium P. aeruginosa.Citation3 Furthermore, in certain cell compartments such as the cytosol of erythrocytes and in lysosomes of other cells PYO is known to accumulate.
Structural analysis of human GR complexed with PYO
Human GR is structurally known in atomic detail.Citation30 As with other disulfide reductases, it contains a so-called drug-binding site in a solvent-filled cavity between the two subunits of the homodimeric enzyme. It is here where heteroaromatic inhibitors such as safranine and xanthene and a number of isoalloxazines and naphtoquinones are bound,Citation28 some of them being sandwiched in an aromate-binding sub-site (see below). The function of this site in vivo is unknown. Using PYO as an inhibitor of crystalline human GR we studied the interactions between protein and ligand in detail.
The structure as determined by x-ray crystallography () shows one PYO molecule bound in the cavity of human GR ( and ). The aromate-binding pocket is formed by seven amino acids (Asn71, Val74, His75, Phe78, Met79, His82, Tyr407) of one subunit and the corresponding residues of the other subunit. PYO is directly sandwiched by residues Val74, Phe78, and their symmetry mates Phe78′ and Val74′.
Table 3. Summary of data collection and structure refinement of the crystalline GR–PYO complex
Superpositions of 461 Cα atoms per subunit of this model with a high resolution structure of human GRCitation30 and with the human GR–xanthene complexCitation31 revealed rms deviations of 0.29 and 0.31 Å, respectively. Differences to the ligand-free structure occur mainly at the positions of the amino acids His75, Phe78, Met79, and His82 and their symmetric counterparts. In the ligand-free structure, these amino acid residues adopt multiple conformations while in the structures with bound PYO or with bound xantheneCitation31 they are rigid and form a defined binding pocket. No remarkable differences were found between the two structures containing an inhibitor. Preliminary crystallographic studies on partially demethylated MB derivatives such as azure B indicate that phenothiazine drugs are also sandwiched between the aromatic rings of phenylalanine 78 and phenylalanine 78′ (Fritz-Wolf, unpublished data). However, the width of the aromate-binding site (about 10 Å) limits the size of tricyclic compounds that can bind in the same orientation as PYO or the aromatic moiety of xanthene. Larger compounds would clash with Asn71 and Tyr407 and their symmetry mates (). These four amino acid residues do not appear to be flexible, because they are involved in multiple interactions with other amino acid residues even in ligand-free GR. PYO is only 7.3 Å long, whereas MB has a length of 12.4 Å and is thus too large to bind in the same way as PYO.
The kinetic finding that PYO can act as a non-competitive inhibitor of human GR suggested that the binding of PYO might lead to major structural changes at the catalytic sites. This, however, did not become evident through the structural analyses. On the other hand, PYO is bound to the basis of the rigid helices in both subunits which carry the catalytic residues Cys58 and Cys63. Very subtle changes at the base of the helices could interfere with the catalytic reduction of GSSG. Another explanation for the lack of observable structural changes is that the ligand exerts its effects only when the substrate glutathione is bound to the crystalline enzyme. This interpretation is consistent with the finding that PYO, as other bi- and tricyclic compounds, can also exhibit uncompetitive inhibitionCitation32 (see above). For testing this, work is in progress to prepare suitable crystals of GR complexed with both the substrate GSSG and the inhibitor PYO.
Effects of PYO on the malaria-causing asexual forms of P. falciparum
The biochemical and crystallographic results strongly suggested systematically studying the antiparasitic activity of PYO in direct comparison with MB. The activity of PYO against chloroquine (CQ)-sensitive and -resistant laboratory strains of P. falciparum was tested as described for MB by Vennerstrom et al.Citation33 and Akoachere et al.Citation17 The IC50 values for PYO were determined to be 86 ± 4.5 nM on the CQ-sensitive strain 3D7 (IC50 for CQ: 8.8 ± 0.9 nM) and 59 ± 3.3 nM on the CQ-resistant K1 strain (IC50 for CQ: 158 ± 12 nM). Thus both parasite strains were about 10–30 times more susceptible to MB than to PYO (IC50 values for of MB: 3.1 ± 0.4 nM for 3D7; 6.6 ± 1.1 nM for K1).
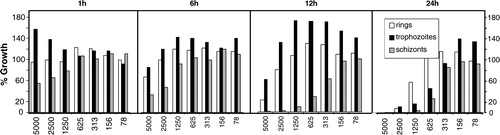
In addition to the IC50 values, we assessed the specificity of PYO action on different developmental stages of intraerythrocytic parasites. For this purpose, seven different PYO concentrations ranging from ∼1 × IC50 to ∼100 × IC50 were added to ring, trophozoite, and schizont stages of 3D7 parasites. After exposure for 1, 6, 12, or 24 hours, PYO was removed and the parasites were incubated for another 24 hours.Citation34 As shown in , PYO showed marked parasiticidal effects already after 1–6 hours of treatment, with schizont stages of the parasites exhibiting the highest susceptibility. For detailed comparison with data on MB and CQ, please consult the papers of Akoachere et al.Citation17 and Sturm et al.Citation34
Figure 5. Stage-specific effects of various concentrations of PYO (x-axis, given in nM) on different developmental stages of the asexual forms of P. falciparum in cultured erythrocytes. The data indicate a rapid antiparasitic effect, with the schizont stage exhibiting the highest susceptibility.
Drug combination assays demonstrated that in vitro PYO acts additively with MB (FIC50 value = 1.0) and synergistically with artemisinin (FIC50 value = 0.73). A synergy with artemisinin and related compounds has also previously been described for MB.Citation17 This supports the notion that the mechanism of antiparasitic action is similar for MB and PYO.
In vivo efficacy and toxicity
In addition to the in vitro studies, PYO and MB were tested in a murine Plasmodium berghei model as described by Peters.Citation35 Preceding in vitro experiments had indicated that P. berghei proliferation is susceptible to PYO with an IC50 of about 115 nM (16 hours incubation with the compound followed by 8 hours incubation with Citation3H-hypoxanthine).
In the first experimental setup for the in vivo testing, the compounds were applied subcutaneously for 3 days after infection. Doses of 3 × 100 mg/kg (n = 4) and 3 × 30 mg/kg (n = 4) MB s.c. were found to be toxic, already leading to death on day 1 (after the first dose). A dosage of 3 × 100 mg/kg PYO s.c. (n = 3) led to rapid death of two animals and to markedly enhanced survival (15 days in comparison with 4-6-7 days for the parasitized non-treated controls) of one animal.
Oral application of 3 × 100 mg/kg MB (n = 4) was found to be very efficient with 0% parasitemia on day 4 (corresponding to 100% activity) and an average mouse survival of 13 days (11, 13, 14, 14 days) in comparison with 4-6-7 days for the parasitized non-treated controls, which were euthanized on day 4. Oral application of 3 × 30 mg/kg MB (n = 4) showed 76.7 ± 5.4% activity on day 4 (parasitemia reduced to 23% of controls) and survival of 14 days for all mice. In direct comparison 3 × 100 mg/kg PYO given orally (n = 2) showed less than 40% activity with respect to parasitemia and no prolonged survival of the animals who additionally suffered from PYO toxicity. In this standard procedure for in vivo testing of antimalarials, treated control animals which are severely ill and about to die 4 days after infection are euthanized on day 4. The efficiency of a drug is expressed by two parameters, the parasitemia and the survival time, which allows one to compare different drugs, concentrations, and combinations with standard clinically employed drugs such as CQ.
In conclusion, MB proved to be a highly efficient and non-toxic antimalarial agent in the P. berghei/mouse model when applied orally in lower doses. In contrast, PYO showed intolerable in vivo toxicity in relation to its antiplasmodial effects in mice.
In vitro activity of MB and PYO against P. falciparum gametocytes
Elimination of blood gametocytes, the sexual form of the parasite, prevents the transmission of disease parasites from man to mosquito and thus interrupts the malaria cycle.Citation36,Citation37 Gametocytes are resistant to most antimalarial drugs except artemisinins and 8-aminoquinolines such as primaquineCitation37,Citation38 and – as indicated by a recent clinical study – to MB.Citation37 In order to complement the clinical study, we systematically investigated the efficacy of MB and PYO on gametocytes in vitro. CQ – still the most used schizonticidal antimalarial drug – was included in these experiments as a reference drug.
MB, PYO and CQ were found to be active against gametocytes of the P. falciparum strain 3D7 on day 9 (stages II and III) and day 13 (stages IV and V). The IC50-values are given in . The day 9 gametocytes were 2, 3, and 50 times more sensitive to MB, PYO, and CQ, respectively, than the day 13 gametocytes.
Table 4. In vitro activities of PYO, MB, and CQ against immature (day 9) and mature (day 13) P. falciparum gametocytes
Discussion
PYO as an important redox-active signaling and antimicrobial molecule is synthesized from chorismate through a series of complex steps.Citation4,Citation39,Citation40 This synthesis is regulated by quorum sensing (for a review see Winstanley et al.Citation41), a process involving a cell-density-dependent accumulation of low molecular weight signal molecules that enable bacteria to modulate the expression of virulence genes and to form biofilms.Citation1–Citation3,Citation42 With its antibiotic function, PYO provides Pseudomonas with comparative growth advantages, which is supported by the fact that, at high cell densities, phenazine biosynthesis was found to be upregulated (up to 300 µM of PYOCitation43). Using a wild-type and a mutant strain defective in phenazine production, Price-Whelan et al.Citation43 studied the physiological role of PYO in Pseudomonas with respect to redox and energy metabolism in more detail. The authors found that the mutant accumulated more NADH in the stationary phase than the wild type which correlated with a decrease in oxygen availability. PYO addition to the phenazine-null mutant decreased intracellular NADH levels, suggesting that PYO reduction facilitates redox balancing. So beside oxygen and nitrate, PYO could be used as an alternative (terminal) electron acceptor. In addition, an inhibition of the pyruvate dehydrogenase complex by PYO through the generation of superoxide has been postulated.Citation43 As a consequence, less pyruvate is converted to acetylCoA for feeding the citric acid cycle and as a result less NADH is produced keeping the NADH/NAD+ ratio in acceptable regions. Additionally to these observations and as delineated in our introduction, a large number of further targets of PYO have been described in Pseudomonas and other organisms – many of them in the context of their redox sensitivity.Citation8,Citation15 In this work we studied the reactivity of PYO and its synthetic analog MB toward important cellular reductants such as NADH, NADPH, or dihydrolipoamide, the reactivity of their reduced leuco-forms toward triplet O2, and their interactions with FAD-containing disulfide reductases in detail.
As shown in , PYO reacts rapidly with NADPH and NADH but is >10 times less reactive than MB toward intracellular dithiol compounds such as thioredoxin or dihydrolipoamide. GSH is a very poor reducing agent for both PYO and MB but the intracellular concentrations of GSH (>1 mM) are one to two orders of magnitude higher than those of other physiological reductants.
However, the reduction of PYO as part of redox cycles can also be brought about by so-called diaphorases (B). These are haem- or flavin-containing enzymes that have narrowly defined physiological functions – e.g. the reduction of disulfides – but as a moonlighter activity they are capable of reducing numerous heteroaromatic compounds at the expense of NADPH or NADH. Taking into account the data from as well as intracellular enzyme concentrations, TrxR and LipDH are likely to be the pharmacologically most relevant PYO-reducing enzymes, since the intracellular concentrations of these enzymes (0.1–1.0 µM) are as high as or even higher than those used for the diaphorase assays.Citation28 Thus, in an example calculated for malaria parasite infected red blood cells, an intracellular concentration of 20 nmol/ml GR is estimated to induce the diaphorase-mediated oxidation of more than 300 µmol NAD(P)H per ml of parasitized cells per day. The corresponding rate for the spontaneous intracellular oxidation of NADPH by PYO is assumed to be about 1% of this value.
The mechanism of the diaphorase activity of flavoenzymes is important for interpreting the action of PYO and MB. Taking human GR as an example, the reducing equivalents are likely to follow a pathway from NADPH via flavin/reduced flavin and PYO/leucoPYO to O2.Citation19 The strongest evidence against an involvement of the active site redox pair Cys58/Cys63 in the diaphorase activity of the disulfide reductases is the finding that mutant proteins which lack one or both active site cysteines have a higher turnover number kcat and a higher catalytic efficiency kcat/KM with PYO as a substrate than the wild-type enzyme; a case in point is the human GR mutant C63A (). In contrast to the diaphorase activity of GR, reduction of the cell-physiological substrate GSSG totally depends on both cysteine residues.
Protein-bound flavin-adenine dinucleotide is known to reduce heteroaromates even when the reaction partners are not precisely oriented to each other.Citation23,Citation44 Regarding the diaphorase activity of GR, a possible binding site of PYO close to reduced flavin is the pocket which accommodates the nicotinamide moiety of NADPH.Citation28,Citation30 In contrast, the binding of PYO in the intersubunit cavity of GR () is unlikely to allow electron transfer from FAD to PYO. The mechanism of the enzymic diaphorase activity will be studied further by structural analyses of crystalline LipDH–PYO complexes. LipDH is preferable to GR as it has a closely related structure but is not inhibited by heteroaromates. The notorious instability of recombinant P. falciparum LipDHCitation45 was overcome by modifications of protein isolation and storage (see Experimental procedures).
The studied disulfide reductases – with the exception of LipDH – are inhibited by heteroaromates like PYO and MB. PYO was found to be a stronger non-competitive inhibitor of human GR than of P. falciparum GR; in contrast MB is a better inhibitor for the parasite enzyme. So far we have not been successful in obtaining crystals of the GR–MB complex that diffract to high resolution. However, in the corresponding GR–PYO complex we were able to identify the binding site of the inhibitor (). This has given us new clues for the binding mode of MB and its demethylated derivatives to disulfide reductases. One hypothesis deduced from our results is that not MB itself but demethylation products of MB might be bound – sandwiched between two symmetrical phenylalanine residues – at the so-called aromate-binding site of GR in vivo.
PYO has been shown to be active against multi-drug-resistant Staphylococcus aureus and against Candida albicans in culture.Citation2 As reported here PYO at nanomolar concentrations is also active against the protozoal parasite P. falciparum in vitro. PYO showed marked parasiticidal effects even after 1–6 hours, with schizont stages of the parasites exhibiting the highest susceptibility. P. falciparum causes more than 300 million cases of clinical malaria per year in tropical regions; groups at risk to develop severe malaria are children under five, pregnant women, and tourists.Citation36 The actual disease is due to growth and multiplication of the parasite in human erythrocytes. The sexual blood forms, so-called gametocytes, do not contribute to clinical disease but are capable of developing in the insect vector thus maintaining the malaria cycle between man and mosquito.Citation37,Citation38 Both PYO and MB show activity against P. falciparum gametocytes in the nanomolar range (). Our systematic in vitro studies furthermore confirm that, in contrast to CQ, MB is indeed highly active against all stages of gametocytes. For the action of CQ on gametocytes, our results were found to be consistent with previous reports.Citation46 The enhanced expression of proteins involved in protection against oxidative stress in mature gametocytes – including glyoxalases, glutaredoxin, and 2-Cys peroxiredoxinCitation47,Citation48 – may contribute to their reduced susceptibility to the tested compounds when compared with asexual red blood cell stages. The fact that MB nevertheless retains great potency against mature gametocytes supports the antimalarial potential of MB – both as a gametocyticidal drug and a drug suitable for schizonticidal drug combinations.Citation37
Because of its toxic effects PYO is not suited as a candidate for antimalarial therapy. Nevertheless it may play a role in vivo. In many wars, Pseudomonas has been responsible for wound infections that give rise to greenish secretions with a typical sweetish odor. Infections in patients with mild and severe forms of cystic fibrosis are also often caused by Pseudomonas species. It remains to be studied to what extent these patients are protected from P. falciparum and P. vivax malaria.Citation49 Further studies on PYO will contribute to our understanding of the biological and cell-biochemical properties of this versatile compound and of its pharmacological analog MB.Citation25,Citation50
Experimental procedures
Enzymes
With the exception of recombinant mitochondrial LipDH of P. falciparum, all enzymes were produced and assayed as described by Buchholz et al.Citation19 The P. falciparum LipDH was cloned, heterologously overexpressed, and purified according to McMillan et al.Citation45 but with the following modifications. To obtain higher yields, we cloned the PCR fragment into pET28 (Novagen, Darmstadt). After induction with IPTG, the cultures were grown over night at 18°C. A one-step purification procedure using Ni-NTA His-Bind resin (Novagen) resulted in more than 10 mg/l cell culture which is 100-fold higher than the previously reported yield.Citation45 For storing and stabilizing the protein according to Blumenstiel et al.,Citation18 P. falciparum LipDH was precipitated with ammonium sulfate and dissolved in a minimum of 100 mM potassium phosphate of pH 7.5. Subsequently, glycerol was added to a final concentration of 30%, and this suspension was stored at −20°C. The enzyme suspension kept its reducing activity versus lipoamide, PYO, and MB for more than 2 months.
Enzyme inhibitors
The concentration of PYO (from Cayman) was determined using an ɛ520 nm of 2.46 mM−1 cm−1 in 0.1 M HClCitation51 or ɛ690 nm = 4.3 mM−1cm−1 in 50 mM MOPS buffer of pH 7.2. 1 mM stock solutions of PYO (in DMSO or in a 50 mM phosphate buffer of pH 8.0) and of MB (in H2O; ɛ613 nm = 40.0 mM−1 cm−1) were freshly prepared at least once a week and kept in dark bottles at room temperature. For biochemical and pharmacological studies the standard buffer of physiological ionic strength (0.3 µ) was 200 mM KCl, 46.5 mM potassium phosphate, 1 mM EDTA, pH 6.9 at 25°C.Citation28,Citation52 The reactions of the various enzymes were performed as described previouslyCitation19 with or without added PYO. In experiments where PYO was tested as a substrate, the spontaneous reaction between NADPH and PYO had to be slowed down. This was achieved by increasing the pH to 8.0. As an alternative one might stabilize the NADPH concentration at less than 10 µM using an NADPH-generating biochemical system.Citation19
Crystallization of human GR
Recombinant human GR was purified as described.Citation52 Crystals were grown at 22°C using the hanging-drop vapor diffusion technique: 10 mg/ml human GR in 120 mM ammonium sulfate, 100 mM potassium phosphate, pH 7.5 was equilibrated by dialysis with 660 mM ammonium sulfate in 100 mM potassium phosphate, pH 7.5. The crystals (>250 µm in each dimension) grew within 1 week. They were washed with 50 mM potassium phosphate of pH 7.3 and soaked in 1 mM PYO in GR assay buffer of pH 7.5 for more than 7 days. Prior to the measurements, the crystals were soaked in mother liquor containing 35% glycerol. Human GR crystals prepared in this way are very stable even in the absence of mother liquor and can be equilibrated with a broad variety of aqueous solutions. This renders human GR crystals uniquely suitable for soaking experiments with pharmacological agents or for using them as indicators of redox potential in living cells.Citation53
X-ray crystallographic studies
X-ray diffraction data were collected at the Max Planck Institute for Medical Research with the rotation method and processed by the program XDS.Citation54 Crystal data were recorded at 100 K by an image plate detector (Mar345 image plate, xrayresearch, Norderstadt, Germany) using CuKα radiation from a rotating anode (Rigaku, MicroMax-007HF, operating at 40 kV and 30 mA). Reflection phasing and refinements were conducted with the PHENIX program suite,Citation55 and model correction was carried out using the graphics program O.Citation56 The structure was solved by molecular replacement, using a high-resolution structure of human GRCitation30 as a search model.
The crystals obey C2 space group symmetry and contain one monomer in the asymmetric unit, with the enzymatically active homodimer generated by a crystallographic two-fold axis. The initial calculated difference map (|Fo|-|Fc|) revealed the presence of electron density resembling PYO. The molecule was found to be bound in the cavity located at the interface of the two GR subunits. The two-fold crystallographic symmetry axis overlaps with PYO, which is not a completely symmetric molecule (A and ); consequently we did all calculations in space group P1, using non-crystallographic symmetry constraints for both subunits.
The inhibitor was placed manually in the additional density. PYO is bound in two orientations, related by a two-fold non-crystallographic axis. Refinement of the full model was completed after a few rounds of model correction (resolution: 2.2 Å, Rfree = 18% with a coordinate error of 0.22 Å and an overall temperature factor of 41.7 ÅCitation2). The model displays good stereochemistry () as verified by Procheck.Citation57
Cultivation of P. falciparum
The CQ-sensitive P. falciparum strain 3D7-Netherlands and the CQ-resistant strain K1 were grown in continuous culture as described by Trager and JensenCitation58 with slight modifications. Unless otherwise stated, parasites were maintained at 1–10% parasitemia and 3.3% hematocrit in an RPMI 1640 culture medium supplemented with A+ erythrocytes, 0.5% lipid-rich bovine serum albumin (Albumax), 9 mM (0.16%) glucose, 0.2 mM hypoxanthine, 2.1 mM l-glutamine, and 22 µg/ml gentamicin. All incubations were carried out at 37°C in 3% O2, 3% CO2, and 94% N2. Synchronization of parasites in culture to ring stages (<70% for drug testing and >90% for stage specificity studies) as starting population was carried out by repetitive treatment with 5% (w/v) sorbitol.Citation59 The parasites were used for the experiments delineated below.
Determination of IC50 values of drugs and fractional inhibitory concentration (FIC) values of drug combinations
Isotopic drug sensitivity assays by means of the semi-automated microdilution techniqueCitation60 were employed to investigate the susceptibility of P. falciparum to various compounds and their effects in combination with other clinically used antimalarials. The procedure depends on the incorporation of radioactive Citation3H-hypoxanthine which is taken up by the parasite as a precursor of purine deoxynucleotides for DNA synthesis and was performed according to the modifications of Fivelman et al.Citation61 In 96-well microtitre plates (NuncR), a two-fold serial dilution of the starting concentration of each pharmacologically active compound to be tested was carried out. Parasites were incubated at a parasitemia of 0.25% (>70% ring forms for the drug testing) and a hematocrit of 1.25% in hypoxanthine-free medium. After 48 hours, 0.5 µCi Citation3H-hypoxanthine was added to each well, and the plates were incubated for another 24 hours. The cells of each well were then harvested on a glass fiber filter (Perkin-Elmer, Rodgau-Jügesheim, Germany), washed and dried. Their radioactivity in counts per minute was assumed to be proportional to the number of parasites in the well. All IC50 values were determined in quadruple.
In order to assess the effects of drug combinations, the two compounds were applied alone and in fixed concentration ratios of 1:1, 1:3, and 3:1 as described by Fivelman et al.Citation61 IC50 and IC90 values were calculated and the FICs of the respective drugs were determined.
Stage-dependent effects of PYO were tested in vitro on synchronous (>90% obtained by repetitive sorbitol synchronization steps) cultures of the P. falciparum strain 3D7 according to Maerki et al.Citation62 For this purpose, seven different PYO concentrations ranging from ∼1 × IC50 to ∼100 × IC50 were added to ring, trophozoite, and schizont stage parasites at a parasitemia of 0.15% and a hematocrit of 5%. After exposure for 1, 6, 12, or 24 hours, the plates were washed four times with compound-free medium in order to dilute the free compound to less than 1% of its starting concentration before adding Citation3H-hypoxanthine and continuing as described above. Compound effects are expressed as a percentage of the untreated controls. Suspensions of uninfected erythrocytes were used for background subtraction. Samples were measured in 96 well plates with 250 µl total volume per well. Typically, the positive controls (untreated) had about 3000 counts per minute (cpm), the negative controls (uninfected erythrocytes) had below 30 cpm. The parasite multiplication rate per red blood cell cycle was in the controls 5.4 + 0.5. For most conditions tested, an initial enhancement of Citation3H-hypoxanthine incorporation was observed before the growth inhibition became apparent. This phenomenon is observed for many drugs and likely to be based on enhanced metabolic activity under pharmacological stress.
In vitro assay for P. falciparum gametocyte sensitivity toward PYO, MB, and CQ
P. falciparum (strain 3D7) gametocytes were cultured according to Ifediba and VanderbergCitation63 with modifications. Briefly, following synchronization with 5% sorbitol,Citation60 a ring stage culture was set up (day 1) with 1.5–2% parasitemia at 6% hematocrit with fresh A+ erythrocytes; no further erythrocytes were used during the experiment. Gametocytes were cultured in RPMI 1640 medium (Gibco) supplemented with A+ human serum (10%), 1.96 g/l glucose, 50 mg/l hypoxanthine, and 22 µg/ml gentamycin. Cultures were kept under 3% O2, 3% CO2, and 94% N2 at 37°C. The medium was changed daily and Giemsa-stained slides were prepared for monitoring gametocyte development. On day 3 the cultures were diluted from 6 to 3% hematocrit. To inhibit further asexual parasite growth,Citation64 cultures were supplemented with 50 mM N-acetylglucosamine (Sigma) from day 5 onwards and then treated with 5% sorbitol on day 8 as described.Citation65 In order to remove hemozoin, the cultures were washed and centrifuged (at 750 g for 10 minutes) four times. For eliminating ring stages and to concentrate gametocytes, the cultures were passed through a magnetically mounted MACS® LS separation column fitted with a 21 G flow resistor (Miltenyi Biotech, Germany) as described.Citation66 Cultures were continued at a 2% gametocytemia and 3% hematocrit until day 9 (stages II and III) or day 13 (stages IV and V) when drug treatment started.
Activity of PYO, MB, and CQ against gametocytes was assessed as described by Chutmongkonkul et al.,Citation67 with modifications. Briefly, stock solutions of CQ diphosphate (Sigma) and MB (Sigma) were freshly dissolved in bidistilled water, PYO was dissolved in DMSO. Twenty microliters of 50% hematocrit (final hematocrit ∼2.0%) with 2% gametocytemia and 500 µl of drug-containing medium or 500 µl solvent-containing medium (for controls) were added to each well in 24-well plates. (Solvent – DMSO or water – refers to the volume of the stock solution used in the respective experiment). After 24 hours, solvent-containing media (controls) and drug-containing media were replaced. After 48 hours all wells received fresh medium without drugs for a further 2 days. On day 14 (for immature gametocytes) and day 18 (for mature gametocytes) thin blood films were prepared, then Giemsa-stained, and gametocytes were counted per 10 000 erythrocytes. The 50% inhibitory concentrations (IC50) were determined by curve fitting the percentage gametocyte inhibition (in relation to control) against log drug concentration with a variable-slope sigmoidal function and compared using the F-test (Prism 4.0 GraphPad Software, San Diego, CA).
Acknowledgments
The authors wish to thank Elisabeth Fischer for her excellent support in cell culture and Timothy Bostick for help with the manuscript. Dr Marion Kerscher, Heidelberg University, kindly confirmed the redox potential of pyocyanin using cyclovoltammetry. We gratefully acknowledge the Deutsche Forschungsgemeinschaft (German Research Foundation) for supporting the study (Grant BE 1540/15-1 to KB and B2 of SFB 544 to RHS). D.K. and S.G. as first authors contributed equally to this paper.
References
- Dietrich LE, Teal TK, Price-Whelan A, Newman DK. Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 2008;321:1203–6.
- Gibson J, Sood A, Hogan DA. Pseudomonas aeruginosa–Candida albicans interactions: localization and fungal toxicity of a phenazine derivative. Appl Environ Microbiol 2009;75:504–13.
- Lau GW, Hassett DJ, Ran H, Kong F. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol Med 2004;10:599–606.
- Mentel M, Ahuja EG, Mavrodi DV, Breinbauer R, Thomashow LS, Blankenfeldt W. Of two make one: the biosynthesis of phenazines. Chembiochem 2009;10:2295–304.
- Wilson R, Sykes DA, Watson D, Rutman A, Taylor GW, Cole PJ. Measurement of Pseudomonas aeruginosa phenazine pigments in sputum and assessment of their contribution to sputum sol toxicity for respiratory epithelium. Infect Immun 1988;56:2515–7.
- O'Malley YQ, Reszka KJ, Spitz DR, Denning GM, Britigan BE. Pseudomonas aeruginosa pyocyanin directly oxidizes glutathione and decreases its levels in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 2004;287:L94–103.
- Muller M. Pyocyanin induces oxidative stress in human endothelial cells and modulates the glutathione redox cycle. Free Radic Biol Med 2002;33:1527–33.
- Ran H, Hassett DJ, Lau GW. Human targets of Pseudomonas aeruginosa pyocyanin. Proc Natl Acad Sci USA 2003;100:14315–20.
- Cheluvappa R, Shimmon R, Dawson M, Hilmer SN, Le Couteur DG. Reactions of Pseudomonas aeruginosa pyocyanin with reduced glutathione. Acta Biochim Pol 2008;55:571–80.
- Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 1999;96:47–56.
- Baron SS, Rowe JJ. Antibiotic action of pyocyanin. Antimicrob Agents Chemother 1981;20:814–20.
- Lau GW, Goumnerov BC, Walendziewicz CL, Hewitson J, Xiao W, Mahajan-Miklos S, et al. The Drosophila melanogaster toll pathway participates in resistance to infection by the gram-negative human pathogen Pseudomonas aeruginosa. Infect Immun 2003;71:4059–66.
- Rahme LG, Ausubel FM, Cao H, Drenkard E, Goumnerov BC, Lau GW, et al. Plants and animals share functionally common bacterial virulence factors. P Natl Acad Sci USA 2000;97:8815–21.
- Cao H, Baldini RL, Rahme LG. Common mechanisms for pathogens of plants and animals. Annu Rev Phytopathol 2001;39:259–84.
- Liu GY, Nizet V. Color me bad: microbial pigments as virulence factors. Trends Microbiol 2009;17:406–13.
- Guttmann P, Ehrlich P. Über die Wirkung des Methylenblau bei Malaria. Berlin Klin Woch 1891;28:953–6.
- Akoachere M, Buchholz K, Fischer E, Burhenne J, Haefeli WE, Schimer RH, et al. In vitro assessment of methylene blue on chloroquine-sensitive and -resistant Plasmodium falciparum strains reveals synergistic action with artemisinins. Antimicrob Agents Chemother 2005;49:4592–7.
- Blumenstiel K, Schoneck R, Yardley V, Croft SL, Krauth-Siegel RL. Nitrofuran drugs as common subversive substrates of Trypanosoma cruzi lipoamide dehydrogenase and trypanothione reductase. Biochem Pharmacol 1999;58:1791–9.
- Buchholz K, Schirmer RH, Eubel JK, Akoachere MB, Dandekar T, Becker K, et al. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob Agents Chemother 2008;52:183–91.
- Meissner P, Adler H, Kasozi D, Fritz-Wolf K, Schirmer RH. The reducing milieu of parasitized cells as a target of antimalarial agents: methylene blue as an ethical drug. In: , Becker K. (ed.) Apicomplexan parasites – molecular approaches toward targeted drug development. Drug discovery in infectious diseases (vol. 2). Weinheim: Wiley-VCH; 2011. p. 115–36.
- Massey V. The identity of diaphorase and lipoyl dehydrogenase. Biochim Biophys Acta 1960;15:314–22.
- Schirmer H. Essay on the medical history of methylene blue [posted 2009 Jul 30]. Available from: http://www.alzforum.org/new/Schirmer.asp.
- Campbell ZT, Baldwin TO. Fre is the major flavin reductase supporting bioluminescence from Vibrio harveyi luciferase in Escherichia coli. J Biol Chem 2009;284:8322–8.
- Baron SS, Terranova G, Rowe JJ. Molecular mechanism of the antimicrobial action of pyocyanin. Curr Microbiol 1989;18:223–30.
- Schirmer RH, Adler H, Zappe HA, Gromer S, Becker K, Coulibaly B, et al. Disulfide reductases as drug targets: methylene blue combination therapies for falciparum malaria in African children. Zaragoza: Prensas Universitarias de Zaragoza; 2008. See http://bzh.db-engine.de/default.asp?lfn=2387.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. USCF Chimera – a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12.
- Färber PM, Arscott LD, Williams CH, Becker K, Schirmer RH. Recombinant Plasmodium falciparum glutathione reductase is inhibited by the antimalarial dye methylene blue. FEBS Lett 1998;422:311–4.
- Krauth-Siegel RL, Bauer H, Schirmer RH. Dithiol proteins as guardians of the intracellular redox milieu in parasites: old and new drug targets in trypanosomes and malaria-causing plasmodia. Angew Chem Int Ed Engl 2005;44:690–715.
- Cornish-Bowden A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem J 1974;137:143–4.
- Berkholz DS, Faber HR, Savvides SN, Karplus PA. Catalytic cycle of human glutathione reductase near 1 A resolution. J Mol Biol 2008;382:371–84.
- Savvides SN, Karplus PA. Kinetics and crystallographic analysis of human glutathione reductase in complex with a xanthene inhibitor. J Biol Chem 1996;271:8101–7.
- Biot C, Bauer H, Schirmer RH, Davioud-Charvet E. 5-substituted tetrazoles as bioisosteres of carboxylic acids. Bioisosterism and mechanistic studies on glutathione reductase inhibitors as antimalarials. J Med Chem 2004;47:5972–83.
- Vennerstrom JL, Makler MT, Angerhofer CK, Williams JA. Antimalarial dyes revisited: xanthenes, azines, oxazines, and thiazines. Antimicrob Agents Chemother 1995;39:2671–7.
- Sturm N, Hu Y, Zimmermann H, Fritz-Wolf K, Wittlin S, Rahlfs S, et al. Compounds structurally related to ellagic acid show improved antiplasmodial activity. Antimicrob Agents Chemother 2009;53:622–30.
- Peters W. Chemotherapy and drug resistance in malaria, vol. 1. London: Academic Press; 1987. p. 542.
- Mendis K, Rietveld A, Warsame M, Bosman A, Greenwood B, Wernsdorfer WH. From malaria control to eradication: The WHO perspective. Trop Med Int Health 2009;14:802–9.
- Coulibaly B, Zoungrana A, Mockenhaupt FP, Schirmer RH, Klose C, Mansmann U, et al. Strong gametocytocidal effect of methylene blue-based combination therapy against falciparum malaria: a randomised controlled trial. PLoS One 2009;4:e5318.
- White NJ. The role of anti-malarial drugs in eliminating malaria. Malar J 2008;7(Suppl 1):S8.
- Mavrodi DV, Bonsall RF, Delaney SM, Soule MJ, Phillips G, Thomashow LS. Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J Bacteriol 2001;183:6454–65.
- Gallagher LA, McKnight SL, Kuznetsova MS, Pesci EC, Manoil C. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J Bacteriol 2002;184:6472–80.
- Winstanley C, Fothergill JL. The role of quorum sensing in chronic cystic fibrosis Pseudomonas aeruginosa infections. FEMS Microbiol Lett 2009;290:1–9.
- Fuqua C, Parsek MR, Greenberg EP. Regulation of gene expression by cell-to-cell communication: acyl-homoserine lactone quorum sensing. Annu Rev Genet 2001;35:439–68.
- Price-Whelan A, Dietrich LE, Newman DK. Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J Bacteriol 2007;189:6372–81.
- Kantz A, Chin F, Nallamothu N, Nguyen T, Gassner GT. Mechanism of flavin transfer and oxygen activation by the two-component flavoenzyme styrene monooxygenase. Arch Biochem Biophys 2005;442:102–16.
- McMillan PJ, Stimmler LM, Foth BJ, McFadden GI, Müller S. The human malaria parasite Plasmodium falciparum possesses two distinct dihydrolipoamide dehydrogenases. Mol Microbiol 2005;55:27–38.
- Smalley ME. Plasmodium falciparum gametocytes: The effect of chloroquine on their development. Trans R Soc Trop Med Hyg 1977;71:526–9.
- Lasonder E, Ishihama Y, Andersen JS, Vermunt AM, Pain A, Sauerwein RW, et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature 2002;419:537–42.
- Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 2003;301:1503–8.
- Shah S. The fever, how malaria has ruled humankind for 500,000 years. New York: Farrar, Straus and Giroux; 2010.
- Zoungrana A, Coulibaly B, Sie A, Walter-Sack I, Mockenhaupt FP, Kouyate B, et al. Safety and efficacy of methylene blue combined with artesunate or amodiaquine for uncomplicated falciparum malaria: a randomized controlled trial from Burkina Faso. PLoS One 2008;3:e1630.
- Reszka KJ, Denning GM, Britigan BE. Photosensitized oxidation and inactivation of pyocyanin, a virulence factor of Pseudomonas aeruginosa. Photochem Photobiol 2006;82:466–73.
- Nordhoff A, Bucheler US, Werner D, Schirmer RH. Folding of the four domains and dimerization are impaired by the Gly446 → Glu exchange in human glutathione reductase. Implications for the design of antiparasitic drugs. Biochemistry 1993;32:4060–6.
- Keese MA, Saffrich R, Dandekar T, Becker K, Schirmer RH. Microinjected glutathione reductase crystals as indicators of the redox status in living cells. FEBS Lett 1999;447:135–8.
- Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 1993;26:795–800.
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 2002;58:1948–54.
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr 1991;A47(Pt 2):110–9.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 1993;26:283–91.
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science 1976;193:673–5.
- Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 1979;65:418–20.
- Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 1979;16:710–8.
- Fivelman QL, Adagu IS, Warhurst DC. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother 2004;48:4097–102.
- Maerki S, Brun R, Charman SA, Dorn A, Matile H, Wittlin S. In vitro assessment of the pharmacodynamic properties and the partitioning of OZ277/RBx-11160 in cultures of Plasmodium falciparum. J Antimicrob Chemother 2006;58:52–8.
- Ifediba T, Vanderberg JP. Complete in vitro maturation of Plasmodium falciparum gametocytes. Nature 1981;294:364–6.
- Ponnudurai T, Lensen AH, Meis JF, Meuwissen JH. Synchronization of Plasmodium falciparum gametocytes using an automated suspension culture system. Parasitol 1986;93(Pt 2):263–74.
- Saul A, Graves P, Edser L. Refractoriness of erythrocytes infected with Plasmodium falciparum gametocytes to lysis by sorbitol. Int J Parasitol 1990;20:1095–7.
- Ribaut C, Berry A, Chevalley S, Reybier K, Morlais I, Parzy D, et al. Concentration and purification by magnetic separation of the erythrocytic stages of all human Plasmodium species. Malar J 2008;7:45.
- Chutmongkonkul M, Maier WA, Seitz HM. A new model for testing gametocytocidal effects of some antimalarial drugs on Plasmodium falciparum in vitro. Ann Trop Med Parasitol 1992;86:207–15.