

Abstract
Objective:
The first oxycodone once daily (OOD) has been developed and after successful pharmacokinetic characterization, therapeutic efficacy and safety were compared to an established oxycodone twice daily (OTD: Oxygesic/OxyContin, Mundipharma).
Design and methods:
A randomized, double-blind, multicenter, cross-over, non-inferiority study was conducted in patients (n = 68) with chronic malignant or non-malignant pain. The new OOD was compared to OTD at identical total daily doses (TDD: 40–120 mg/day) employing intensive, five times daily current pain (0–100 mm visual analog scale, VAS) and twice daily 12 h recalled pain assessments as well as safety parameters such as nausea and sedation (VAS) over 5 days for each treatment (after a 5 day run-in phase).
Results:
There was no significant difference in analgesic potency detected between the two treatments based on 95% CI for difference in the daily mean current pain (−2.09 mm VAS) over 5 days, determined as −5.09 to 0.91 mm VAS. A difference ≤12 mm VAS indicated non-inferiority of OOD, i.e. lack of clinically relevant difference in analgesia. Intake of rescue medication had no effect on study results as evaluated by ANCOVA. The difference in adverse events (AEs) between the two treatments did not reach significance, as 19.1% and 23.5% of patients experienced treatment-related AEs while on OOD and OTD, respectively. Advantages for OOD regarding consistency of analgesia (i.e. use of rescue medication, current and recalled pain) and sedation did not reach statistical significance in this limited study population.
Conclusion:
Despite the small number of patients and short study duration, the results support the conclusion that new OOD is (at least) equivalent to established OTD regarding safety and efficacy.
Introduction
Chronic pain is a condition with major impact on quality of life and resulting in a high level of disability. The World Health Organization (WHO) estimated that only non-malignant pain from low back pain, osteoarthritis and rheumatoid arthritis resulted in the loss of more than 22 million disability-adjusted life years in the year 2002Citation1. Nowadays the recommendations on the use of strong opioids according to the WHO analgesic ladder for the treatment of cancer pain are implemented successfully world-wide in developed and developing countriesCitation2, with 70% of chronic-pain patients in Europe being managed in primary careCitation3.
Oxycodone is one of the most frequently used opioids in the management of moderate to severe chronic pain and its use is on the rise world-wide, as it has analgesic and safety properties comparable to morphine while at the same time exhibiting higher and more consistent per oral bioavailabilityCitation4–6. The use of prolonged release formulations is an established approach to avoid strong fluctuations of opioid levels over the day and during the night for obtaining stable analgesiaCitation7. However, compliance with the analgesic regimens is considered to be only around 40% in oncology outpatients and an important obstacle for obtaining adequate analgesia. Also patients with chronic non-malignant pain tend to use analgesics at lower doses and dosing frequency than prescribed, with treatment complexity representing one of the reasons for this non-compliance to the recommended dosing regimenCitation8. Therefore improving compliance and patient satisfaction is a constant target in clinical management of chronic pain, both in cancer and non-cancer patients. Reduction in dosing frequency to once daily administration is an established approach to meet these targets, even in pain managementCitation6,Citation9,Citation10; compliance has been found to be significantly higher for once daily formulations compared to four times daily regimens (79% vs. 51%, p < 0.001)Citation11. Although hydromorphoneCitation8 as well as morphineCitation12 are available as once daily formulations, the more widely used oxycodone has not so far been available as such a favorable formulation.
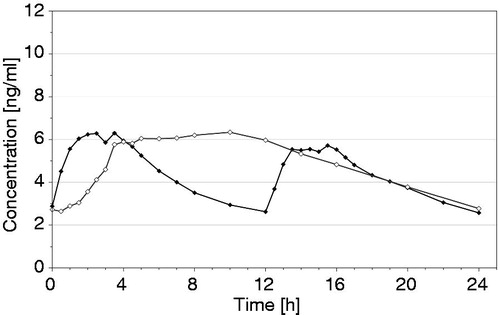
Consequently the first oxycodone once daily (OOD) has been developed. Initially, the OOD has been characterized for its pharmacokinetic properties (PK) compared to the established oxycodone twice daily (OTD) confirming bioequivalence after single (fasted/fed conditions) and multiple dosing in the relevant PK parameters AUC, Cmax and Cmin (publication on PK in preparation)Citation13,Citation14. Overall, the oxycodone plasma concentration–time profile resulting from the new OOD shows the typical pattern of a once daily formulation with a gradual increase during the first 4–6 hours followed by a plateau maintained for approximately 10 hours and a slow decline (). The new OOD formulation is designed to have abuse deterrent properties regarding expected methods of abuse (crushing, heating, snorting and i.v. application) and to have alcohol resistant properties regarding misuse by concomitant alcohol consumption.
Figure 1. Mean (arithmetic mean) plasma concentration–time curves of oxycodone at steady state after oral administration of oxycodone 10 mg once daily (test, ◊) or oxycodone 5 mg twice daily (reference, ♦) under fasting conditions in 36 healthy subjects (for details see ScheidelCitation13).
In the following, the clinical evaluation of the analgesic potency for this new OOD formulation in a non-inferiority trial employing an efficient cross-over design is described. In general for analgesic non-inferiority trials, large patient populations and long treatment periods are necessary when using parallel-group study designsCitation15–17; however, in the past there have been successful trials employing smaller patient populations and shorter treatment intervals in well controlled cross-over studiesCitation18–21.
Methods
This was a prospective, multicenter, randomized, double-blind, active-controlled, adaptive, two-treatment, two-period, two-sequence, cross-over study (EudraCT 2010-020402-15). The primary objective of the study was to demonstrate non-inferiority of the newly developed oxycodone once daily (OOD) prolonged release tablet compared with established twice daily oxycodone (OTD) prolonged release tablet at the same total daily dose (TDD) in patients with chronic malignant and non-malignant pain. The TDD of oxycodone as determined during a titration/stabilization phase was fixed throughout the study. Pain assessment was based on the current pain rated by the patient on a 0 to 100 mm visual analog scale (VAS) assessed five times daily throughout the two 10 day double-blind treatment periods. Secondary objectives were the assessment of safety and tolerability of the new OOD in comparison with OTD.
The sample size for the study was estimated based on power simulations, assuming a difference in the primary efficacy parameter (mean current pain on VAS) between OOD and OTD of 0 mm, a standard deviation for this parameter in the range of 10 to 24 mm and correlation of the two treatment periods of 0 to 0.9. A difference of 12 mm on VAS was considered the largest clinically acceptable difference between the two products. Based on these assumptions, it could be shown by simulations that a power of 80% to 90% can be reached for a non-inferiority trial by including ≤60 patients in almost all scenarios simulated. Including potential drop-outs from the per protocol population, the study was planned with an initial sample size of 76 patients (38 per treatment sequence) and the possibility to adjust the sample size via an unblinded interim analysis (see Statistical analysis).
The treatment sequence (test–reference; reference–test) was assigned by randomization code and central randomization procedure.
Patient population
Caucasian males and females aged ≥18 years were recruited into the study if they had a documented history of chronic malignant or non-malignant, predominantly non-neuropathic pain (as assessed by the DN4 Neuropathic Pain Diagnostic QuestionnaireCitation22) requiring at least 40 mg oxycodone (or equivalent) per day; an Eastern Cooperative Oncology Group (ECOG) performance status <3Citation23 and a life expectancy of at least 3 months.
Exclusion criteria included history of hypersensitivity to oxycodone or any excipient of the study medication; requirement of >120 mg oxycodone per day (or equivalent), surgery within 1 month prior to start and/or anticipated or scheduled surgical intervention during study; intravenous chemotherapy and/or radiotherapy for pain alleviation and/or neural blockade within 2 weeks prior to study start; significant hepatic impairment or severe renal impairment (creatinine clearance <30 ml/min); females who were pregnant or lactating.
At the end of a titration/stabilization phase and prior to randomization adequate analgesia (daily mean current pain ≤40 mm on VAS) and analgesic requirements (≥40 mg oxycodone per day with ≤2 doses of rescue medication (10 mg immediate release morphine sulfate) had to be stable for each of three consecutive days.
Study design
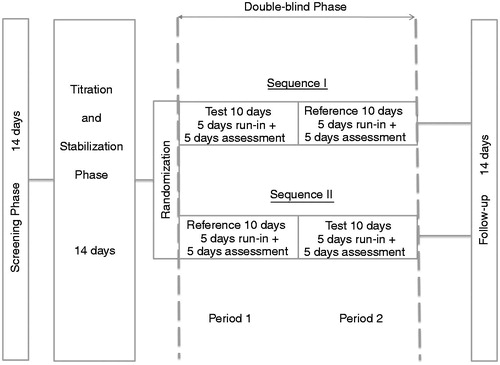
The study consisted of a screening phase and a subsequent titration/stabilization phase (maximum 14 days each), followed by a double-blind treatment phase in which the patients received the two treatments by one of the two sequences, i.e. sequence I (test = OOD, followed by reference = OTD, i.e. Oxygesic, Mundipharma, Germany) or sequence II (reference–test), each medication given for 10 days (). During the titration/stabilization period the patients were switched to oxycodone and dose was adjusted for sufficient pain relief, i.e. daily mean current pain ≤40 mm on VAS. After this phase all patients with stable pain control, stable analgesic requirements and satisfying all criteria for inclusion and exclusion predefined in the study protocol were randomized to enter the double-blind treatment phase of the study. After 10 days of treatment with the first study medication of the respective sequence, patients were directly switched to the second medication without wash-out. No dose adjustment for oxycodone was allowed throughout the double-blind treatment phase; rescue medication (10 mg immediate release morphine sulfate) was supplied to patients.
Figure 2. Study design for testing non-inferiority of oxycodone once daily (test) versus oxycodone twice daily (reference) at identical total daily doses.
OTD, OOD and a dummy were blinded using the same type of over-encapsulation. The patients received the same number of encapsulated tablets in both periods of the double-blind treatment phase in the morning (OTD or OOD) and in the evening (OTD or dummy).
Efficacy assessment
Data on the efficacy of the study medication was collected in daily diaries throughout the titration/stabilization phase as well as during intake of study medication. Current pain was assessed by the patient on a 0 to 100 mm VAS five times daily at 08:00 h (before intake of study medication), 11:00 h, 14:00 h, 17:00 h and 20:00 h (before intake of study medication). For all assessments a maximum deviation of ±20 minutes from the prespecified time points was allowed. At all these time points, patients evaluated the current pain, and at 08:00 h and 20:00 h they also evaluated the recalled pain over the past 12 h (recalled pain during day- and night-time). Throughout the titration/stabilization phase the daily mean of the five current pain assessments had to be ≤40 mm VAS for each of three consecutive days before randomization for the study medication. During the double-blind phase of the study, for each study medication the current pain and recalled pain scores obtained from days 6 to 10 were employed for statistical evaluations. Days 1 to 5 of each period were regarded as an active 5 day run-in phase in order to avoid any potential carry-over effects between the different study periods.
For each patient the daily mean current pain intensity for the last 5 days of the test and reference treatment period was calculated, besides the mean current pain for each day and for the different time points over these 5 days (i.e. mean current pain at 08:00 h, 11:00 h, 14:00 h, 17:00 h and 20:00 h over these 5 days). For the recalled pain over the last 12 h, the mean over the last 5 days of each treatment period was calculated.
The overall effectiveness (pain control) of the medication over the treatment periods was additionally assessed by patient and investigator on a 0 to 3 categorical scale (CAT; 0 = not effective, 3 = highly effective) and the use of rescue medication was recorded throughout titration as well as treatment periods.
Safety assessment
Safety assessment consisted of monitoring and recording all adverse events (AEs) and serious adverse events (SAEs) using MedDRA (version 10.1) as well as monitoring of hematology, blood chemistry, urine parameters, vital signs and performing physical examinations. Special focus was on patients’ self-assessment (daily diaries) of nausea and sedation, assessed by using independent 0 to 100 mm VAS at 08:00 h and 20:00 h.
Statistical analysis
The full analysis population (FA) consisted of all patients with at least one measurement of the primary efficacy parameter. Patients dropping out prior to start of the second treatment period could not be analyzed for efficacy and were excluded from this subset. For drop-outs during the second treatment period, patients were included/excluded for efficacy analysis on case-by-case decision performed during blinded data review meetings. The per-protocol (PP) population included the patients without any major protocol deviations. For safety evaluation all randomized patients were included who received at least one dose of study medication (safety data set).
Analysis of variance (ANOVA) was performed for the primary efficacy data including the factors center, sequence, patient × sequence, period and treatment. For primary efficacy evaluation, non-inferiority of test vs. reference medication was tested by calculating the two-sided 95% confidence interval (CI) for the difference in daily mean current pain (VAS) over the last 5 days of each treatment period. Non-inferiority of test medication was concluded if the upper limit of this 95% CI calculated from residual standard error obtained from ANOVA did not exceed 12 mm. If the 95% CI for this parameter did not exceed 0, it was planned to claim superiority of test medication over reference. Moreover, the primary efficacy parameter was evaluated including the use of rescue medication as a covariate in the analysis of variance (ANCOVA). The secondary pain intensity parameters obtained (except the 0–3 CAT) were evaluated employing the same statistical model, while the 0–3 CAT was evaluated with non-parametric Wilcoxon test adjusted for the cross-over design. The primary efficacy analysis was performed for the PP data set as this preserves a conservative decision when using non-inferiority testing, analyses for the FA population were performed to demonstrate robustness of the trial results. Secondary efficacy parameters were generally calculated for the FA. All statistics were performed employing SAS (version 9).
An adaptive, unblinded interim analysisCitation24 was prospectively planned after completing 24 patients in the PP data set, in order to obtain an estimate for the variability underlying the initial sample-size estimation and potential subsequent adaptation of sample size or early termination of study due to success or inefficacy. The method employed was based on Fisher’s combination test and controlled the overall significance level of the test decision. The interim analysis was performed by an independent data monitoring committee (IDMC) consisting of two independent statisticians and one further medical expert. All persons otherwise involved in the conduct of the trial remained blinded.
The patient incidence and number of reports of treatment-related AEs were calculated and presented for each medication using MedDRA system organ class and preferred term. Absolute values and changes to baseline of vital signs and laboratory parameters were analyzed using descriptive statistics.
Results
Eighty-nine patients were screened, of these 85 entered into the titration/stabilization phase, 71 were finally randomized and 68 took at least one dose of study medication (safety data set). Sixty patients were in the full analysis (FA) data set (70.6%, 29 for sequence I and 31 for sequence II) and 56 completed the trial (27 sequence I and 29 sequence II) of whom 46 qualified for the per protocol (PP) data set (23 per sequence). Patients with malignant pain (n = 40) had tumors with primary localization in the lung (7), breast (6), cervix (5), prostate (5), colon/rectum/anus (4), oropharynx (3), skin (2), lymphoma (2) or other localizations (6), respectively. Non-malignant pain (n = 20) was associated with lower back pain/cervical spine associated conditions (14), chronic arthritis (3), chronic knee/hip pain (2) and one case of chronic pain in elbow related to contusion. The main reason for withdrawal was failure to attain adequate and stable analgesia during titration phase (n = 7) or adverse events (n = 14), while only two patients withdrew due to lack of treatment efficacy, two due to other screening failure, and one each due to non-compliance with the study protocol or withdrawal of consent due to personal reasons.
Mean score obtained from the DN4 Neuropathic Pain Diagnostic Questionnaire at screening was 1.2 (range 0 to 3) in the safety data set, indicating likely nociceptive pain in all patients included. The FA data set included 66.7% of patients with malignant pain. The daily mean current pain scores on the last 3 days of the titration/stabilization phase were 15.2, 15.6 and 14.8 mm VAS, respectively (FA data set). All patients kept the oxycodone doses determined during titration phase constant throughout the two 10 day double-blind treatment periods. Compliance based on case report forms was 99.3% of the test and 99.5% of the reference medication. In the FA data set (n = 60), 23 patients received a total daily dose (TDD) of 40 mg oxycodone, 15 patients 60 mg, 10 patients 80 mg, 4 patients 100 mg and 8 patients 120 mg. There were no differences in patient demographics between the treatment sequences (). The cross-over design of the study inherently avoided any potential imbalances between treatment groups and allowed for an intra-individual comparison of the efficacy of both study medications.
Table 1. Patient demographics at baseline for sequence I (test–reference), sequence II (reference–test) and total (safety) data set.
Efficacy
Primary endpoint
The primary objective of the study was to demonstrate non-inferiority of the new oxycodone once daily formulation (OOD = test) in comparison to the established oxycodone twice daily formulation (OTD = reference) at identical total daily dose (TDD) based on the daily mean for current pain assessed five times daily over the last 5 days of each treatment period on 0 to 100 mm VAS for the PP population (n = 46). Over the 5 day evaluation period the mean current pain score was 18.78 mm VAS for OOD and 20.87 mm VAS for OTD () i.e. the difference between test and reference was −2.09 mm VAS. The two-sided 95% CI for the difference was −5.09 mm to 0.91 mm VAS (). As the limits of the 95% CI did not exceed the prespecified margin of 12 mm VAS, non-inferiority of the test medication was concluded.
Table 2. Primary efficacy outcome, mean current pain (mm on 0 to 100 mm VAS) over the last 5 days of each treatment period for total as well as for period 1 and 2 of the cross-over, double-blind treatment phase of the study in the per protocol data set (n = 46).
Table 3. Statistical evaluation of primary efficacy parameter, difference in mean current pain (mm VAS on 0 to 100 mm VAS) over the last 5 days of each treatment period between oxycodone once daily (T) and oxycodone twice daily (R) at equal total daily doses, for complete population as well as subpopulations in different study parts (at/after interim analysis, part 1/2) and for different types of pain (malignant, non-malignant) in the per protocol data set (n = 46).
The planned interim analysis for the results of the first 24 patients in the PP population (part 1) had concluded non-inferiority of the test medication, therefore patient recruitment was stopped and another 22 patients already included finished the study in the PP population (part 2). The results obtained in both parts of the study (at/after interim analysis) independently confirmed non-inferiority of the test medication, with 95% CI for difference in mean current pain of −5.03 to 2.21 mm VAS for part 1 and −8.79 to 1.72 mm VAS for part 2 (). Although patients with non-malignant pain in general exhibited higher current pain scores (test: 29.9 ± 21.3 mm VAS, reference: 33.6 ± 21.7, n = 15) than patients with malignant pain (test: 13.4 ± 10.1, reference: 14.7 ± 10.7, n = 31), statistical evaluation confirmed non-inferiority of test medication in both subpopulations independently (). Moreover, results obtained in the FA data set entirely confirmed the results obtained in the PP data set (data not shown).
Secondary endpoints
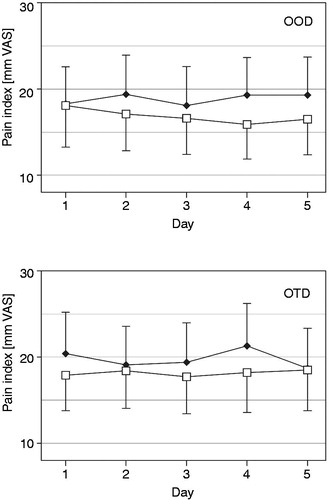
The daily means of the five assessments of current pain have been evaluated separately for each of the last 5 days of test and reference medication. There was no clinically relevant fluctuation (>12 mm VAS) of the mean current pain, both for the FA as well as for the PP population (). Also for the five daily current pain evaluations (08:00 h, 11:00 h, 14:00 h, 17:00 h and 20:00 h) no statistically significant fluctuations over time of day or between days were observed. Representative data for the evaluations at 08:00 h and 20:00 h are depicted in , indicating a trend for lower pain in the morning than in the evening (both treatments) as well as a trend to more consistent analgesia with test in the evening (not significant [n.s.]). The patients also scored the recalled pain over the last 12 h each day at 08:00 h and 20:00 h; the mean for this parameter over the last five treatment days was 17.5 and 19.2 mm VAS for test as well as 20.1 and 20.7 mm VAS for reference, respectively (n.s.). On the 4 point categorical scale (0–3 CAT) 90.0% of the investigator’s ratings and 86.7% of the patient’s ratings assessed the test medication as moderately or highly effective, compared to 86.6% and 81.7%, respectively, for the reference medication (n.s.).
Figure 3. Daily mean current pain (mm on 0 to 100 mm VAS, mean with 95% CI) over the last five treatment days for oxycodone once daily (□) and oxycodone twice daily (•) at identical total daily doses in the full analysis data set (n = 60).
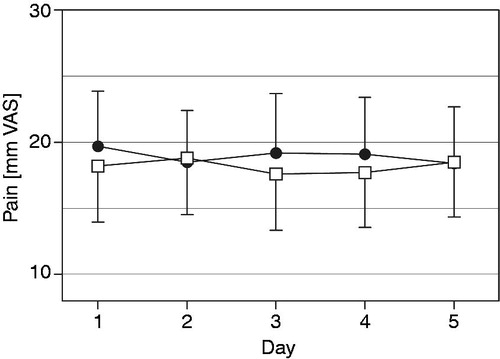
Figure 4. Current pain (mm on 0 to 100 mm VAS, mean with 95% CI) over the last five treatment days for oxycodone once daily (OOD) and oxycodone twice daily (OTD) at identical total daily doses for two of the five daily time points of pain evaluation (□ = evaluated at 08:00 h,♦ = evaluated at 20:00 h) in the full analysis data set (n = 60).
Moreover the use of immediate-release morphine supplied to the patients as rescue medication has been analyzed. During the blinded data review discrepancies between use of rescue medication as documented in patient diary and the amount of returned rescue medication were observed. Therefore statistical evaluation was performed for both sources of documentation. Including the amount of rescue medication as a covariate in the statistical evaluation of the mean current pain resulted in no significant effect on the study outcome, either for the PP or for the FA population (data not shown). The mean daily dose of rescue medication over the last 5 days of the treatment period was 9.1 mg morphine for the test and 11.2 mg for the reference treatment. No significant difference in the use of rescue medication was observed between the two study medications or the two treatment periods; however, for the reference treatment there was a trend for higher cumulative doses over the last 5 days of the second period compared to the first period (40.0 mg vs. 72.8 mg), while no such effect was observed for the new OOD (43.8 mg vs. 47.4 mg) in the FA population.
Safety
In general the incidence of adverse events (AEs) was similar for both treatments with a total of 39.7% and 38.2% of patients treated experiencing AEs with OOD and OTD, respectively, in the safety data set (n = 68). The number of patients reporting AEs related to study medication was lower for OOD, i.e. 19.1% vs. 23.5%, respectively; however, this difference did not reach statistical significance. None of the 18 serious AEs reported in 16 patients were considered to be related to study medication. Two deaths occurred throughout the treatment period, both related to underlying malignant disease, none suspected to be related to study medication. Four patients discontinued treatment due to an AE, thereof two after the end of the treatment phase. Most frequently typical gastrointestinal side-effects of opioid therapy were reported, such as nausea, constipation and vomiting (). Two cases of dyspnea were considered to be treatment related.
Table 4. Number of subjects (n and %) as well as incidence (nAE) of adverse events by system organ class (≥4.4%) and preferred term (>1.5%) in the safety data set (n = 68).
The analysis of laboratory parameters did not disclose any obvious clinically relevant trends. A serious case of pancytopenia with unlikely relationship to the test medication was reported after the double-blind treatment phase. One patient experienced moderate hypoglycemia during the titration and stabilization phase, which was reported as an AE. The patient had a history of insulin-dependent diabetes. There were no apparent clinically relevant trends with regard to vital signs (systolic and diastolic pressure, pulse rate, and body temperature) and no evidence of respiratory depression (respiratory rate measured at pre-defined time points) during the study. The study did not disclose any so far unknown safety signals for the newly developed oxycodone once daily formulation.
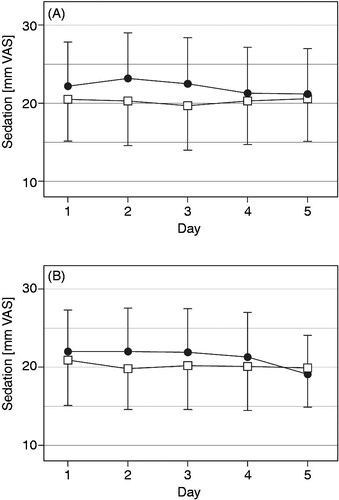
As nausea and sedation are well known side effects of opioid therapy in general, these effects were monitored twice daily employing a 0 to 100 mm VAS. The mean intensity of nausea ranged between 3.7 and 5.5 mm on the VAS throughout the 10 days of each treatment period for the test medication and between 3.7 and 7.5 for the reference medication (n.s.). No significant differences between the two medications were observed within the treatment period (). For the patients’ sedation in the morning and evening of each treatment day there was a trend towards lower scores for OOD (, n.s.).
Figure 5. Nausea (mm on 0 to 100 mm VAS, mean with 95% CI) in the morning (A) and evening (B) over the last five treatment days for oxycodone once daily (□) and oxycodone twice daily (•) at identical total daily doses in the safety data set (n = 68).
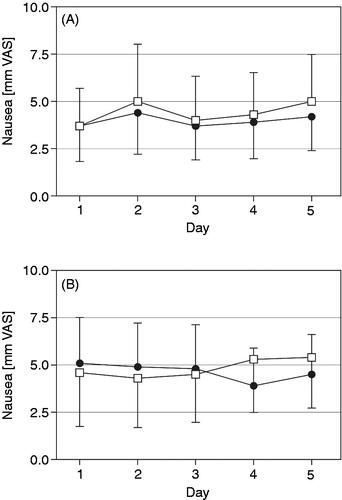
Figure 6. Sedation (mm on 0 to 100 mm VAS, mean with 95% CI) in the morning (A) and evening (B) over the last five treatment days for oxycodone once daily (□) and oxycodone twice daily (•) at identical total daily doses in the safety data set (n = 68).
Discussion
All results obtained from this clinical study in patients with malignant and non-malignant pain demonstrate that the newly developed oxycodone once daily (OOD) prolonged release tablet provides analgesia at least equally effective compared to established oxycodone twice daily (OTD) prolonged release tablets regarding both safety and efficacy. Non-inferiority of the OOD treatment to OTD could be established for the primary target parameter in both the per protocol and the full analysis populations. Switching between the two medications based on identical total daily dose for oxycodone provided reliable and safe analgesia over 24 h. The study was terminated early as a preplanned interim analysis confirmed non-inferiority of OOD regarding the primary target parameter of efficacy. Moreover, the results obtained for the secondary efficacy parameters, including recalled pain over the last 12 h as well as categorical assessment of analgesic efficacy by investigator and patient clearly indicate that the prolonged release characteristics described for OOD translate to an analgesic performance equivalent to OTD in clinical practice. Regarding specific safety parameters for opioid analgesia assessing nausea and sedation, both treatment options performed comparably, with slight advantages observed for OOD regarding sedation scores, an effect also observed for consistency of analgesia over the day, which, however, did not reach statistical significance in either case.
The somewhat lower incidence of break-through pain and reduced need for rescue medication with OOD (n.s.) are in line with the established pharmacokinetic profile of the new OOD (), exhibiting only one trough in daily oxycodone plasma concentration in the morning, the time of day with overall lowest pain scores assessed, and avoiding low blood levels in the evening, when frequently the highest pain scores are observed. Positive effects of this lack of evening trough can be observed for the 20:00 h pain scores obtained (), with OOD resulting in consistent mean scores below 20 mm VAS, while slightly higher and more variable scores were obtained with OTD; however, these differences did not reached statistical significance in the limited number of patients necessary to meet the primary clinical endpoint. Even at the time of trough plasma concentrations with the OOD in the morning, adequate analgesia was observed according to patients’ self-assessed pain scores. Therefore the OOD provides stable and reliable clinical effect to the patient at least equal to the established OTD. Close monitoring of pain status by five times daily patient assessment in the trial presented here allowed for this differentiated evaluation of efficacy of both treatments.
Adverse events reported in the study were expected considering the established pharmacological profile of oxycodone, especially since a large fraction of patients of the safety dataset (46 of 68) suffered from malignant pain. The majority of treatment-related adverse events are well known side effects of opioid analgesics and there was no significant difference between the test and the reference product. Nausea scores as assessed by patients were overall low and exhibited no significant or relevant differences between the two study medications tested, while scores for sedation under OOD tended to be slightly better than for OTD, albeit never reaching statistical significance.
The cross-over design employed could be shown to be highly effective to meet the targets of this trial, as proposed in recent recommendations on the design of clinical trials on analgesics, indicating the usefulness of cross-over studies for this purposeCitation25. In patients with chronic pain wash-out phases between treatments have to be considered inadequate, due to ethical reasons. Therefore a 5 day run-in phase before assessment of efficacy of each treatment was part of the study design. Potential carry-over between the two assessment phases can be considered to be marginal based on the fact that the 5 day run-in is long enough to avoid any pharmacokinetic overlap between the formulations employed, exhibiting terminal half-lives in the range of 6 to 7 h for oxycodone (publication on PK in preparation); moreover, the patient × sequence effect in the ANOVA employed did not reveal any significant carry-over effect. Finally the evaluation of the study results stratified for period () gave no hint of such an effect, excluding the presence of any kind of ‘pharmacological’ carry-over between the treatments, e.g. via alterations at the level of receptors or transporters.
Pain assessment by the patient via VAS is considered a valid and reliable approach in clinical practice and for scientific purposesCitation26; however, consensus for statistical evaluation of this important clinical parameter is apparently not fully adopted yet. Even recently studies have been published comparing pain scores employing purely statistical means (t-test) defining an arbitrary level of significance as the cut-off for a meaningful difference, without evaluating the clinical relevance of potential differences detected by this methodologyCitation27. Though it is nowadays widely accepted that treatment-related differences observed with pain assessment tools have to be evaluated employing a threshold based on clinically relevant differences between treatments, rather than purely statistical significances. Currently there is no overall consensus on which difference in VAS represents this cut-off for clinically relevant differences in pain intensity. Published clinical trials considered a difference of at least 17 mmCitation18,Citation20, 18–20 mmCitation28,Citation29 or 25 mm as relevantCitation21,Citation30. A recent review on the use of VAS for chronic pain assessment found typically differences from 10 to 40 mm VAS as the threshold for clinically significant differences between treatmentsCitation31. Others reported a change in pain intensity of 33% as clinically relevantCitation32, while the IMMPACT IV meeting recommended a change of 30% as a meaningful pain intensity change only when the absolute pain intensity score is >3 on the 0 to 10 numerical rating scale (NRS), equivalent to 30 mm VASCitation33, e.g. comparing the effect of analgesics to previously untreated condition or placebo treatmentCitation34; otherwise a chance of 10 to 20 mm was considered to make a relevant difference for the patientCitation35. For the study described here a conservative approach was employed, considering a difference in pain intensity of ≤12 mm VAS to indicate non-inferiority of the new OOD formulation.
Conclusion
The data presented here fully support the conclusion that the newly developed oxycodone once daily prolonged release tablet is (at least) equal to the established oxycodone twice daily prolonged release tablet regarding both analgesia assessed by patient and physician, and clinical safety in patients with moderate to severe malignant and non-malignant pain. The study population was selected for patients with low-grade neuropathic pain. Although there are many positive reports on the clinical efficacy of oxycodone in neuropathic painCitation36–38, it is generally accepted that, although analgesic effects are evident, the use of opioids for neuropathic pain is mostly second- or third-line therapyCitation38. Even more importantly for this trial, neuropathic pain is associated by frequent pain crises, providing a less stable baseline for assessment of analgesic efficacyCitation39,Citation40. Therefore the inclusion of patients with low neuropathic pain scores in the trial presented here is justified by the special nature of this condition in clinical pain management and the requirement of stable clinical pain assessment conditions for non-inferiority trials in general.
Transparency
Declaration of funding
The study was funded by study sponsor Develco Pharma Schweiz AG.
Declaration of financial/other relationships
M.A.M. has disclosed that she is an employee of the study sponsor, Develco Pharma Schweiz AG. E.A.L. and M.J. have disclosed that they participated in the study as clinical investigators; E.A.L. was also coordinating investigator. They both received investigator fees from Develco Pharma Schweiz AG.
CMRO peer reviewer 1 has no relevant financial or other relationships to disclose. Peer reviewer 2 has disclosed that he is a consultant to Grunenthal and Johnson & Johnson; and that he is on the Speakers’ Bureau of Grunenthal, Mundipharma and Pfizer.
Acknowledgments
The authors thank the investigators from all 12 study sites in Germany and Poland for taking part in this study – Drs. Lux, Hafer, Schütze, Schwittay, Strick in Germany and Drs. Janecki, Cialkowska-Rysz, Chemperek-Wroczek, Korozan, Lembas-Sznabel, Sokalszczuk, Stachowiak in Poland. The sponsor thanks Dr. Gudrun Nold for valuable input to the study design as well as Dr. Andreas Völp, Prof. Dr. Gerd Geisslinger and Per Settergren Sørensen for their contributions as members of the independent data monitoring committee. Harrison Clinical Research GmbH (now SynteractHCR Deutschland GmbH), Munich, Germany, provided services as Clinical Research Organisation (CRO) including data management and statistical evaluation. The authors also thank Dr. Stephan Döppenschmitt for providing editorial support, which was also funded by Develco Pharma Schweiz AG.
References
- WHO. Treatment Guidelines on Chronic Non-Malignant Pain in Adults. 2008. Available at: http://www.who.int/medicines/areas/quality_safety/Scoping_WHOGuide_non-malignant_pain_adults.pdf [Last accessed 1 February 2014]
- WHO. Normative Guidelines on Pain Management. Report of a Delphi Study to Determine the Need for Guidelines and to Identify the Number and Topics of Guidelines That Should Be Developed by WHO. 2007. Available at: http://www.who.int/medicines/areas/quality_safety/delphi_study_pain_guidelines.pdf [Last accessed 1 February 2014]
- Johnson M, Collett B, Castro-Lopes JM. The challenges of pain management in primary care: a pan-European survey. J Pain Res 2013;6:393-401
- Thapa D, Rastogi V, Ahuja V. Cancer pain management – current status. J Anaesthesiol Clin Pharmacol 2011;27:162-8
- Pergolizzi J, Boger RH, Budd K, et al. Opioids and the management of chronic severe pain in the elderly: consensus statement of an International Expert Panel with focus on the six clinically most often used World Health Organization Step III opioids (buprenorphine, fentanyl, hydromorphone, methadone, morphine, oxycodone). Pain Pract 2008;8:287-313
- Levy MH. Advancement of opioid analgesia with controlled-release oxycodone. Eur J Pain 2001;5(Suppl A):113-16
- Coluzzi F, Mattia C. Chronic non-cancer pain: focus on once-daily tramadol formulations. Ther Clin Risk Manag 2007;3:819-29
- Coluzzi F, Mattia C. OROS(R) hydromorphone in chronic pain management: when drug delivery technology matches clinical needs. Minerva Anestesiol 2010;76:1072-84
- Cramer MP, Saks SR. Translating safety, efficacy and compliance into economic value for controlled release dosage forms. Pharmacoeconomics 1994;5:482-504
- Stambaugh JE, Reder RF, Stambaugh MD, et al. Double-blind, randomized comparison of the analgesic and pharmacokinetic profiles of controlled- and immediate-release oral oxycodone in cancer pain patients. J Clin Pharmacol 2001;41:500-6
- Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther 2001;23:1296-310
- Panjabi SS, Panjabi RS, Shepherd MD, et al. Extended-release, once-daily morphine (Avinza) for the treatment of chronic nonmalignant pain: effect on pain, depressive symptoms, and cognition. Pain Med 2008;9:985-93
- Data on file. Comparative bioavailability studies of oxycodone after single/multiple oral administration of Oxycodone HCl 10 mg PR tablets XL (Develco Pharma Schweiz AG) and single/multiple oral administration two times daily of Oxygesic® 5 mg tablets (Mundipharma GmbH, Germany) under fasting/fed conditions in healthy subjects. Clinical Study Reports by Scheidel B et al., 683B8, 684B8, and 685B8 of 28-Apr-2010
- Data on file. Dose proportionality studies of oxycodone after single oral administration of Oxycodone HCl 10 mg PR tablets XL, Oxycodone HCl 20 mg PR tablets XL,Oxycodone HCl 40 mg PR tablets XL, and Oxycodone HCl 80 mg PR tablets XL (Develco Pharma Schweiz AG) under fasting conditions in healthy subjects. Clinical Study Reports by Scheidel B et al., 686B8 of 22-Oct-2010 and 265B12 of 22-Nov-2012
- Bingham CO 3rd, Sebba AI, Rubin BR, et al. Efficacy and safety of etoricoxib 30 mg and celecoxib 200 mg in the treatment of osteoarthritis in two identically designed, randomized, placebo-controlled, non-inferiority studies. Rheumatology (Oxford) 2007;46:496-507
- Fardellone P, Zaim M, Saurel AS, Maheu E. Comparative efficacy and safety study of two chondroitin sulfate preparations from different origin (avian and bovine) in symptomatic osteoarthritis of the knee. Open Rheumatol J 2013;7:1-12
- Hale ME, Ahdieh H, Ma T, Rauck R. Efficacy and safety of OPANA ER (oxymorphone extended release) for relief of moderate to severe chronic low back pain in opioid-experienced patients: a 12-week, randomized, double-blind, placebo-controlled study. J Pain 2007;8:175-84
- Hagen NA, Babul N. Comparative clinical efficacy and safety of a novel controlled-release oxycodone formulation and controlled-release hydromorphone in the treatment of cancer pain. Cancer 1997;79:1428-37
- Grosset AB, Roberts MS, Woodson ME, et al. Comparative efficacy of oral extended-release hydromorphone and immediate-release hydromorphone in patients with persistent moderate to severe pain: two randomized controlled trials. J Pain Symptom Manage 2005;29:584-94
- Bruera E, Belzile M, Pituskin E, et al. Randomized, double-blind, cross-over trial comparing safety and efficacy of oral controlled-release oxycodone with controlled-release morphine in patients with cancer pain. J Clin Oncol 1998;16:3222-9
- Finn JW, Walsh TD, MacDonald N, et al. Placebo-blinded study of morphine sulfate sustained-release tablets and immediate-release morphine sulfate solution in outpatients with chronic pain due to advanced cancer. J Clin Oncol 1993;11:967-72
- Bouhassira D, Attal N, Alchaar H, et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain 2005;114:29-36
- Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982;5:649-55
- Bauer P, Kohne K. Evaluation of experiments with adaptive interim analyses. Biometrics 1994;50:1029-41
- Dworkin RH, Turk DC, Peirce-Sandner S, et al. Considerations for improving assay sensitivity in chronic pain clinical trials: IMMPACT recommendations. Pain 2012;153:1148-58
- Jensen MP. The Validity and Reliability of Pain Measures for use in Clinical Trials in Adults: Review Paper Written for the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) meeting, 12–13 April 2003. Available at: http://www.immpact.org/static/meetings/Immpact2/background/Jensen_review.pdf [Last accessed 1 February 2014]
- Hsu SK, Yeh CC, Lin CJ, Hsieh YJ. An open label trial of the effects and safety profile of extended-release tramadol in the management of chronic pain. Acta Anaesthesiol Taiwan 2012;50:101-5
- Hagg O, Fritzell P, Nordwall A. The clinical importance of changes in outcome scores after treatment for chronic low back pain. Eur Spine J 2003;12:12-20
- Grilo RM, Treves R, Preux PM, et al. Clinically relevant VAS pain score change in patients with acute rheumatic conditions. Joint Bone Spine 2007;74:358-61
- Walsh TD, MacDonald N, Bruera E, et al. A controlled study of sustained-release morphine sulfate tablets in chronic pain from advanced cancer. Am J Clin Oncol 1992;15:268-72
- Ruyssen-Witrand A, Tuback F, Ravaud P. Systematic review reveals heterogeneity in definition of a clinically relevant difference in pain. J Clin Epidemiol 2011;64:463-70
- Coluzzi PH, Schwartzberg L, Conroy JD, et al. Breakthrough cancer pain: a randomized trial comparing oral transmucosal fentanyl citrate (OTFC) and morphine sulfate immediate release (MSIR). Pain 2001;91:123-30
- Jensen MP. Meaningful change and meaningful endpoints in pain clinical trials. IMMPACT IV meeting. 2004. Available at: http://www.immpact.org/meetings/Immpact4/background4.html [Last accessed 1 February 2014]
- Peloso PM, Gammaitoni A, Smugar SS, et al. Longitudinal numbers-needed-to-treat (NNT) for achieving various levels of analgesic response and improvement with etoricoxib, naproxen, and placebo in ankylosing spondylitis. BMC Musculoskelet Disord 2011;12:165
- Dworkin RH, Turk DC, Wyrwich KW, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain 2008;9:105-21
- Watson CP, Moulin D, Watt-Watson J, et al. Controlled-release oxycodone relieves neuropathic pain: a randomized controlled trial in painful diabetic neuropathy. Pain 2003;105:71-8
- Gimbel JS, Richards P, Portenoy RK. Controlled-release oxycodone for pain in diabetic neuropathy: a randomized controlled trial. Neurology 2003;60:927-34
- McNicol ED, Midbari A, Eisenberg E. Opioids for neuropathic pain. Cochrane Database Syst Rev 2013;8:CD006146
- Gatti A, Sabato AF, Occhioni R, et al. Controlled-release oxycodone and pregabalin in the treatment of neuropathic pain: results of a multicenter Italian study. Eur Neurol 2009;61:129-37
- Dworkin RH, Barbano RL, Tyring SK, et al. A randomized, placebo-controlled trial of oxycodone and of gabapentin for acute pain in herpes zoster. Pain 2009;142:209-17