
Abstract
Background The quality of biologicals, including biosimilars, is subject to change as a result of manufacturing process modifications following initial authorization. It is important that such product changes have no adverse impact on product efficacy or safety, including immunogenicity.
Objectives The aim of this study was to investigate the number and types of manufacturing changes for originator mAbs (the reference for the comparability exercise to confirm biosimilarity) according to European Public Assessment Report (EPAR) documentation and to ascertain the level of risk these changes might impart. The extensive body of evidence contained in the EPAR documents can help support the EMA during the EC marketing authorization approval process for biosimilars, since it provides a broad base of scientific experience.
Research designs and methods For EPAR-listed mAbs, details of all changes listed chronologically in the EPAR were evaluated and described. Based on these descriptions the manufacturing changes can be categorized by risk status (low, moderate or high).
Results Entries for 29 mAbs with publicly available EPAR reports were reviewed. These contained details of 404 manufacturing changes authorized by the European Medicines Agency (EMA): 22 were categorized as high risk, 286 as moderate risk and 96 as low risk manufacturing changes. A limitation of this analysis is that it only summarizes publicly available data from EPAR documents.
Conclusions Manufacturing change data indicate that the EMA has significant experience of process changes for originator mAbs, and the impact they may have on the efficacy and safety of biologicals. This experience will be useful in biosimilar product development to ensure adherence to sound scientific principles. Compared with the established manufacturing process for a reference product, the production of biosimilars will usually be different. Consequently, in addition to a comprehensive comparative functional and physicochemical characterization analysis, clinical data is required to confirm mAb biosimilarity.
Introduction
It has been estimated that more than 250 biologicals derived from recombinant DNA technology have been approved for medicinal use in humansCitation1. The high cost of these agents has created a barrier to their routine use. The introduction of biosimilar medicinal products (biosimilars), non-identical but highly similar copies of previously approved biological therapeutics (referred to as the reference products), provides a method of improving access to these drugs in a more cost-effective mannerCitation2. This might facilitate wider usage in a greater number of individuals who could benefit from such treatments. However, biological agents are generally large complex proteins that are difficult to characterize and difficult to copy. Differences in structure between the biosimilar and reference product, small or large, may impact the properties of the biosimilarCitation3. The European Medicines Agency (EMA) has developed a regulatory framework for the development of biosimilars to ensure that they are highly similar to the reference product, in terms of structure and function, pharmacodynamics and mechanism of action, pharmacokinetic properties, and clinical efficacy and safety (including immunogenicity)Citation4. A landmark in biopharmaceutical research was achieved in September 2013 when, based on the scientific recommendation of the EMA, the EC granted marketing authorizations for two biosimilar infliximab monoclonal antibodies (mAbs) for use in the same indications as the originator reference infliximab productCitation5,Citation6. The Food and Drug Administration (FDA) in the United States has issued draft guidelines for the development of biosimilarsCitation7, and in March 2015 ZarxioFootnote*(filgrastim-sndz) was the first biosimilar product to be approved in the United StatesCitation8.
Approval of the first biosimilar agents in Europe and the United States has brought the issue of variability of biological medicines to the attention of practicing clinicians. The quality attributes of all biotechnology products are subject to change as a result of manufacturing process modifications following the initial authorization. This applies to reference products as well as to biosimilarsCitation3. According to the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use guideline (ICH Q5E), a comparability exercise should provide analytical evidence that, based on appropriate comparison of relevant quality attributes, pre- and post-change products are highly similar and considered comparable. Such product changes should have no adverse impact on product efficacy or safety, including immunogenicityCitation9. The comparability exercise has become the scientific norm for biosimilar development, and is based on a combination of chemical and analytical testing, biological assays, and, in some cases, additional non-clinical and clinical (safety and/or efficacy) dataCitation3. In a review of biosimilars development Schneider presented details of the number of manufacturing changes after authorization for 11 mAbs and fusion proteins (e.g. etanercept) approved for use in rheumatology indications. The number of changes ranged from 0 (rilonacept) to 37 (infliximab) with a median of four. Three products had in excess of 15 changes in manufacturing (infliximab, etanercept and rituximab)Citation3. The documented changes could be as simple as a change of address of the authorization holder to more complex changes in the manufacturing process.
The aim of the current analysis was to update the findings of Schneider in relation to mAbs, and to collect and thoroughly investigate the number and types of manufacturing changes for originator mAbs according to their European Public Assessment Report (EPAR) documentationCitation10. The EMA’s EPAR documents provide a broad level of scientific experience which can be used in the future to evaluate process changes for biological drugs (including biosimilars) so that the necessary comparability tests can be applied to help obtain EC marketing authorization approval.
Methods
In October 2014 publicly available EPARs for all mAbs authorized by the EMA between 1998 and October 2014 were analyzedCitation10. Details of all changes listed chronologically in the ‘Procedural steps taken and scientific information after the authorisation’ section were evaluated and categorized by risk status (low, moderate or high), as defined by Lee et al. ()Citation1.
Figure 1. Manufacturing changes for biotechnology products were categorized as low/moderate/high riskCitation1. Adapted from Lee et al. Comparability and biosimilarity: considerations for the healthcare provider. CMRO 2012;28:1053-8.

Below we include examples of statements from EPARs registered with the EMA. The changes were assessed by regulatory experts and, depending on the significance of the change, they were rated as low, moderate or high risk. The same change (e.g. change in manufacturer) can be rated at different risk levels depending on the significance of the change. The following published (EPAR) examples are categorized as high-risk statusCitation1,Citation10:
Change in the purification of the active substance.
Change in synthesis or recovery of a non-pharmacopoeial or novel excipient.
Change in the manufacturer of AS (active substance) or of a starting material/reagent/intermediate for AS – the proposed manufacturer uses a substantially different route of synthesis or manufacturing conditions.
Change in batch size (including batch size ranges) of AS or intermediate – the change requires assessment of the comparability of a biological/immunological AS.
Change to in-process tests or limits applied during the manufacture of the AS – widening of the approved in-process test limits, which may have a significant effect on the overall quality of the AS.
We categorized from the EPAR documents the following examples as moderate-risk manufacturing changesCitation1,Citation10:
Change(s) to the manufacturing process for the active substance.
Changes in the manufacturing process of the AS – the change refers to a [-] substance in the manufacture of a biological/immunological medicinal product and is not related to a protocol.
Change in the manufacturer of AS or of a starting material/reagent/intermediate for AS.
Change to in-process tests or limits applied during the manufacture of the AS.
Change in the manufacturer of AS or of a starting material/reagent/intermediate for AS – the change relates to a biological AS or a starting material [-] used in the manufacture of a biological/immunological product.
Low-risk manufacturing changes are based on the following EPAR examplesCitation1,Citation10:
Change in the manufacturing process of the finished product (FP).
Replacement or addition of a manufacturing site for the FP – site where any manufacturing operation(s) take place, except batch release, batch control, and secondary packaging, for biological/immunological medicinal products.
Change to in-process tests or limits applied during the manufacture of the finished product.
Change in immediate packaging of the finished product – qualitative and quantitative composition.
Changes to the manufacturing facility of the finished product.
Results
A total of 36 entries for mAbs were found published on the EMA website and 29 of these had publicly available EPAR reports (). As of October 2014, these 29 EPAR reports contained details of 404 manufacturing changes authorized by the EMA. Of these, 22 were categorized as high-risk, 286 as moderate-risk and 96 as low-risk manufacturing changes (). The number of changes ranged from 0 (pertuzumab) to 50 (infliximab) with a median of 11 per product, and five products had in excess of 25 changes in manufacturing (infliximab, basiliximab, trastuzumab, adalimumab and eculizumab) (). Since the elapsed time after authorization differs significantly (1998–2013) the annual number of manufacturing changes was introduced for comparison. The annual average number was 1.8 (range 0–3.71) ().
Figure 2. Number of manufacturing changes for monoclonal antibodies in their European Public Assessment Reports according to risk category (during the search period all non-proprietary names relate only to the trade named medicines listed in ).
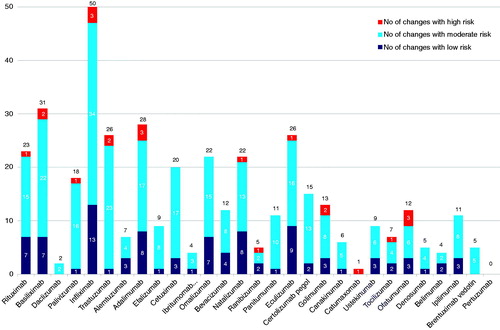
Figure 3. Average number of manufacturing changes per product year elapsed since registration (during the search period all non-proprietary names relate only to the trade named medicines listed in ).
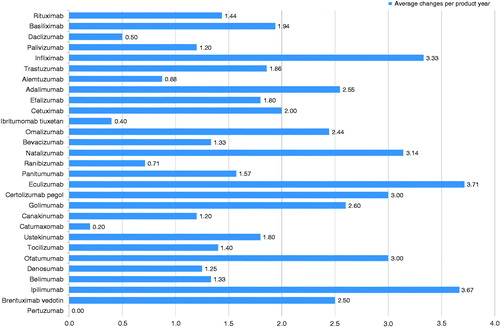
Table 1. Monoclonal antibody medicines and their EPARTable Footnote* ‘Procedural steps…’ documents from EMATable Footnote** webpage during the search periodCitation10.
Discussion
Biological products vary over time as a result of modifications to the manufacturing process. Consequently, manufacturers need to demonstrate the consistency and robustness of the manufacturing process by implementing good manufacturing practices, modern quality control and assurance procedures, in-process controls and process validationCitation11. This applies to both originator products and biosimilars, and the manufacturing process for a biosimilar should be optimized to minimize differences between it and its reference protein. As noted in this short review, the regulatory authorities have extensive experience of assessing manufacturing changes over time and their impact on the efficacy and safety of the final product, for a wide range of mAbs. These same principles, using state-of-the-art science and technology, are being applied to biosimilar manufacturing and quality control.
The scientific principles for assessing manufacturing process changes of mAbs are well established, and are based upon comprehensive and thorough physicochemical, analytical and functional comparative assessments to ensure comparability. These principles are encompassed in the ICH comparability guidance document ICH Q5E which has been used by the EMA and other regulatory bodies to develop their comparability guidelinesCitation9. This guideline defines the rationale and background for accepting changes in the production of biological products and acknowledges that… “the demonstration of comparability does not necessarily mean that the quality attributes of the pre-change and post-change product are identical, but that they are highly similar and that the existing knowledge is sufficiently predictive to ensure that any differences in quality attributes have no adverse impact upon safety or efficacy of the drug product.” This applies to both reference proteins and biosimilars. Interestingly, Schiestl and colleagues investigated the variation in quality of different batches of three marketed biological therapeutics (darbepoetin alfa, rituximab and etanercept) and found differences in the glycosylation profiles of all three productsCitation12. The originators’ batch variants were predicted not to alter the clinical profiles of any drugs, and these were acceptable to the health authorities and all three products remained on the market.
Trust in the development of biosimilars is founded on the expertise of the regulatory authorities which has been gained from their experience with originator biologicals. Indeed, the entire biosimilar development process is built on a solid foundation of extensive analytical and physicochemical characterization which is robustly assessed. This is supported by the accumulated experience of the EMA over the last couple of decades from the scientific evaluation of manufacturing changes of originator biologicals and their impact on clinical outcomes.
The findings of this study are limited to publicly available data from the EU regulatory authority’s documents for mAbs for the time period 1998 to October 2014 (). Since the manufacturing processes of biologics are continuously changing during their life-time, monitoring and assessing the changes will provide examples of change consequences in the production process which can help guide future decisions. It should be noted that no information of process changes for biosimilar mAbs was available up to October 2014.
Conclusions
The manufacturing change data presented herein indicate that, prior to the authorization of the first biosimilar mAb, the EMA had extensively evaluated the manufacturing process changes of originator mAbs, and gained significant experience in the change process and its impact on the safety and efficacy of biologicals. These comparability exercises became the guiding principles of biosimilarity to confirm no meaningful differences in quality, safety and efficacy. These exercises have been employed in biosimilar product development to ensure that sound scientific principles are adhered to. Since the manufacturing process for biosimilars will likely be different from the reference product for proprietary reasonsCitation1, physicians ought to be able to trust the expertise of regulatory authorities to confirm the similarity of previously approved originator products and their biosimilars akin to the pre- and post-manufacturing changes of original biologicals.
Transparency
Declaration of funding
This paper was funded by Egis Pharmaceuticals plc, Budapest, Hungary.
Declaration of financial/other relationships
B.V., Z.B., M.S., and Z.Z. have disclosed that, during the publication process, they were employees of Egis Pharmaceuticals plc.
CMRO peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Notice of correction
Please note that Figure 1, 2, 3 and Table 1 have been corrected since the article was first published online (25 February 2016).
Acknowledgments
The authors thank Dr Steve Clissold of Content Ed Net for editorial support.
Previous presentation: Parts of this paper were presented at 35th European Workshop for Rheumatology Research, 5–7 March 2015, Budapest, HungaryCitation13.
References
- Lee JF, Litten JB, Grampp G. Comparability and biosimilarity: considerations for the healthcare provider. Curr Med Res Opin 2012;28:1053–8
- Bennett CL, Chen B, Hermanson T, et al. Regulatory and clinical considerations for biosimilar oncology drugs. Lancet Oncol 2014;15:e594–605
- Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis 2013;72:315–18
- European Medicines Agency. Guideline on similar biological medicinal products, CHMP/437/04 Rev 1. Available at: http://www.ema.europa.eu/ema/pages/includes/document/open_document.jsp?webContentId=WC500176768 [Last accessed November 2015]
- European Medicines Agency. European Medicines Agency recommends approval of first two monoclonal antibody biosimilars. 2013. Available at: www.ema.europa.eu/docs/en_GB/document_library/Press_release/2013/06/WC500144941.pdf. [Last accessed January 2016]
- Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe. mAbs 2013;5:621–3
- Alten R, Cronstein BN. Clinical trial development for biosimilars. Semin Arthritis Rheum 2015;44:S2–8
- FDA Press Announcement/ucm436648, March 6, 2015. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm [Last accessed September 2015]
- International Conference on Harmonisation (ICH) of Technical Requirements for registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process Q5E. 2004. Available at: www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf [Last accessed July 2015]
- European Medicines Agency. European public assessment reports. Available at: www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d125 [Last accessed July 2015]
- World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs) 2009. Available at: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf [Last accessed January 2016]
- Schiestl M, Stangler T, Torella C, et al. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol 2011;29:310–12
- Zrubka Zs, Vezer B, Sebeszta M, Authorised manufacturing changes of therapeutic monoclonal antibodies in EPAR documents. Poster A8.31 at 23th European Workshop for Rheumatology Research, 5–7 March 2015, Budapest, Hungary