Abstract
At present, 150 clinical trials are registered with the National Cancer Institute, which investigate the efficacy of inhibitors of the PI3K/Akt/mTOR pathway against multiple cancers. Efficacy varies not so much with drug action, but with tumor type, as different cancer types (and different pre-clinical models) exhibit widely differing susceptibilities to mTOR inhibitors, such as rapamycin. Viral cancers appear to be among the most mTOR-addicted and most rapamycin-sensitive cancers. We discuss the different mTOR inhibitors that are currently available and in clinical trials. We also speculate how the molecular makeup of viral cancers could guide the selection and use of known and novel mTOR inhibitors to treat virus-associated malignancies.
The term ‘Rapalogs’ describes a class of allosteric inhibitors that target the mammalian target of rapamycin (mTOR) pathway. More recently, ATP-competitive inhibitors of mTOR as well as so-called ‘dual-kinase’ inhibitors have been developed, which target other kinases in the mTOR signaling cascade in addition to mTOR itself. Rapamycin (sirolimus) was discovered in the 1970s, and is in widespread use as a second-generation oral immune suppressant in solid organ transplantation. Rapamycin inhibits IL-2 translation and secretion in T cells and thus T cell proliferation (). In addition, it also inhibits IL-2-dependent (and other ligand)-dependent signaling in the same cells. In this context, the cell-autonomous G1 arrest phenotype induced by protein translation arrest is augmented by inhibition of IL-2, which is a paracrine and autocrine growth factor for T cells. The first-generation immune suppressants, cyclosporine and FK506, also inhibit IL-2 expression in T cells and thereby T cell proliferation. However, their inhibition is T cell specific, because the inhibitory mechanism ultimately depends on NFAT (nuclear factor of activated T cells), a T cell lineage-restricted transcriptional transactivator of the IL-2 promoter. By contrast, rapalogs inhibit the ubiquitously required mTOR kinase and thereby inhibit protein translation in all cell types, including cancer cells.
Figure 1. Model of rapamycin modes of action in transplantation (left) and cancer (right). If used as immune suppressants in solid organ transplantation, both rapamycin and FK506 inhibit translation of essential cytokines for activated T cells (IL-2). Rapamycin also inhibits the translation of essential cytokines for activated B cells (IL-6). If used as anti-cancer drugs for viral cancers, both rapamycin and FK506 inhibit IL-2 in herpesvirus saimiri (HVS)-induced T cell lymphoma (TL). Rapamycin also inhibits IL-6 in KSHV-induced primary effusion lymphoma (PEL). Eventually, clones of TL and PEL evolve, which no longer depend on IL-6 or in which IL-6 expression is rapamycin insensitive Citation[3].
![Figure 1. Model of rapamycin modes of action in transplantation (left) and cancer (right). If used as immune suppressants in solid organ transplantation, both rapamycin and FK506 inhibit translation of essential cytokines for activated T cells (IL-2). Rapamycin also inhibits the translation of essential cytokines for activated B cells (IL-6). If used as anti-cancer drugs for viral cancers, both rapamycin and FK506 inhibit IL-2 in herpesvirus saimiri (HVS)-induced T cell lymphoma (TL). Rapamycin also inhibits IL-6 in KSHV-induced primary effusion lymphoma (PEL). Eventually, clones of TL and PEL evolve, which no longer depend on IL-6 or in which IL-6 expression is rapamycin insensitive Citation[3].](/cms/asset/c782f573-51c8-46ad-8069-07876a0c5438/ieid_a_642369_f0001_b.jpg)
Rapamycin is ‘tumorstatic’ rather than ‘tumortoxic’ because mTOR controls protein synthesis and volume growth rather than DNA replication-driven cell proliferation. This mechanism of action limits rapamycin's potency as an anti-cancer agent, except in those cancers where mTOR does not just regulate translation in general, but regulates translation of specific autocrine-acting cytokines required for cancer cell survival. Virus-associated cancers (predominantly herpesvirus-associated B and T cell lymphomas) are examples of this tumor class. Here, rapalogs display nanomolar IC50s in cell culture and in pre-clinical models Citation[1-5]. The efficacy of rapalogs against other subtypes of cancer have been observed in clinical trials, notably in sarcomas, mantle cell lymphoma and renal cell carcinoma, and most dramatically in Kaposi sarcoma (KS), which is associated with human herpesvirus 8 or Kaposi sarcoma-associated herpesvirus (KSHV).
In transplant-associated KS, switching from the immunosuppressant drug cyclosporine A to the immunosuppressant drug rapamycin (sirolimus) resulted in resolution of cutaneous KS Citation[6]. All tumor lesions disappeared but graft function did not decline. This study thus separated rapamycin's immunosuppressive function (on T cells) from its anti-cancer effects on the endothelial lineage tumor KS. Since then, similar results have been reported by others Citation[7,8], although exceptions have been noted as well Citation[9]. Discordant case studies are part of the norm, particularly in a highly pre-treated patient population. This should not detract from the general mechanism. A randomized clinical trial to formally establish the efficacy of any rapalog against KS is still missing.
KS tumor cells are firmly addicted to mTOR signaling. KS lesions are characterized molecularly by high-level phosphorylation of Akt, mTOR and the mTORC1 targets, p70 S6 kinase and ribosomal protein S6 Citation[6,10,11]. In other systems, rapamycin blocked focus formation induced by oncogenic alleles of PI3K or of Akt Citation[12]. These observations place mTOR downstream of, and epistatic to, PI3K and Akt.
Modern mTOR inhibitors promise to improve on the clinical efficacy of rapamycin in several ways. The first class of modern mTOR inhibitors or rapalogs are allosteric inhibitors of mTORC1. They display better bioavailability and pharmacokinetics than sirolimus, but they follow the same molecular mechanism. Everolimus, temsirolimus and ridaforolimus form a mTORC1:FKBP:rapalog complex analogous to rapamycin (sirolimus). Prior binding to FKBP is required and mTORC1 is the direct target of inhibition; a second complex, mTORC2, is not affected. The interactions are a bit more complicated, since the same catalytic subunit of mTOR kinase participates in both mTORC1 and mTORC2 complexes, with specificity being contributed by different co-factors (raptor in case of mTORC1 and rictor in case of mTORC2). Depending on tumor type and stoichiometry, this can lead to opposing indirect effects: sequestering mTOR kinases into an inactive mTORC1:FKBP:rapalog complex may deplete mTOR and thus diminish mTORC2 kinase activity as well; alternatively mTORC2 activity may increase and in effect compensate for loss of mTORC1 through feedback activation of Akt Citation[13]. Which scenario ultimately prevails is cell type-specific and may help explain the wide range of rapamycin responsiveness among different tumors.
Direct clinical comparisons among the rapalogs have not been reported, but in our animal studies, all allosteric-acting rapalogs exhibit equivalent efficacy across a wide range of tumor xenograft models (unpublished observation); cell lines/tumor types that exhibit a high IC50 against rapamycin tend to also be partially resistant to allosteric-acting rapalogs.
The second class of modern mTOR inhibitors target the active site of mTOR kinase (Torin/PP242/Ku-0063794/WYE-354) and in some cases have been shown to inhibit tumor cells more effectively than rapamycin Citation[14]. It appears that active site mTOR inhibitors have weaker effects on the proliferation and function of normal lymphocytes than rapalogs. Additionally, one would expect active site inhibitors to be associated with the rapid development of resistance as well as correspondingly greater potential for dose-limiting side effects than rapalogs, which are allosteric inhibitors.
Some active site mTOR inhibitors (e.g., NVP-BEZ335, PI-103) also inhibit PI3K. Dual inhibitors like NVP-BEZ235 are being tested in clinical trials () (reviewed in Citation[15]). These tend to be associated with greater efficacy as well as a wider range of susceptible tumor types based on pre-clinical experiments Citation[1,2,14,16-18]. The exact molecular mechanism for this increased potency probably lies in the expanded range of kinase targets that are inhibited. Inhibiting PI3K affects many more targets (e.g., mTOR-independent targets of Akt). Additionally, PI3K inhibition results in Akt inhibition as well. Consequently, mTORC2-driven Akt activation (due to mTORC1 inhibition-induced feedback) should also be suppressed. At present, the biological significance of only a few targets of mTORC1 and mTORC2 has been established. A very recent study combined an allosteric rapalog with a dual PI3K/mTOR active site inhibitor Citation[14,19], which resulted in additive efficacy. It remains to be seen if the increased tumor efficacy of dual inhibitors is associated with increased suppression of normal lymphocyte proliferation.
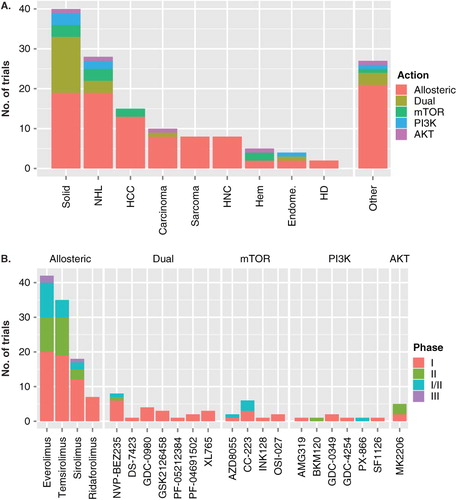
Figure 2. Current NCI registered clinical trials using inhibitors of the PI3K/Akt/mTOR pathway. (A). Stacked bar chart showing the number of trials (on the vertical axis) for different cancer categories (on the horizontal axis). The class of inhibitor is indicated by color. NHL, non-Hodgkin lymphoma; HCC, any liver cancer including hepatocellular carcinoma; HNC, head and neck cancer; Hem, hematopoietic malignancies including multicentric Castleman disease and leukemia; endome., endometrial cancer; HD, Hodgkin disease. (B) Stacked bar chart showing the number of trials (on the vertical axis) for different mTOR inhibitors (on the horizontal axis) grouped by mechanism of action. The phase of the trial is indicated by color.
Recently, a new molecular phenotype of mTOR inhibitors has been uncovered. Inhibition of mTOR either by allosteric or active site inhibitors, as well as the dual PI3K/mTOR inhibitors has been shown to induce autophagy Citation[20,21]. In cancer cells, autophagy is one mechanism used by tumor cells to survive anti-neoplastic chemotherapies. Interestingly, although rapamycin by itself could induce autophagy in glioma cells, competitive mTOR inhibitors like Ku-0063794 induced autophagy to a greater degree than rapamycin. Chloroquine is an inhibitor of autophagy and the combination of chloroquine with dual inhibitors of PI3K and mTOR was found to effectively induce apoptosis in glioma Citation[21]. Thus, the addition of autophagy inhibitors to PI3K/mTOR inhibitors may be warranted in the future.
Currently, extensive safety studies on active site mTOR inhibitors or PI3K/mTOR dual inhibitors have not been reported. Hence, our predictions of clinical utility are highly speculative. Several questions remain to be answered. Should we prioritize individualized therapy and find out when, where and why the established, safe allosteric rapalogs work? Or should we bet on the power of combinatorial therapy, assuming that dual PI3K/mTOR inhibitors will be inherently more efficacious? What is the safety profile when combination inhibitors are added to the dual inhibitors? In this era of targeted therapies, targeting multiple pathways critical for tumor cell survival is warranted. However, as we target more pathways, the potential for side effects and cytotoxicity is greatly increased and the safety profiles of such combinations need to be considered in equal proportion to their efficacy.
Declaration of interest
This work was supported by NIH (CA163217 and CA096500). The authors have no other competing interests to declare. The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Bibliography
- Bhatt AP, Bhende PM, Sin SH, Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood 2010;21(22):4455-63
- Chaisuparat R, Hu J, Jham BC, Dual inhibition of PI3Kalpha and mTOR as an alternative treatment for Kaposi's sarcoma. Cancer Res 2008;68(20):8361-8
- Sin SH, Roy D, Wang L, Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood 2007;109(5):2165-73
- Wlodarski P, Kasprzycka M, Liu X, Activation of mammalian target of rapamycin in transformed B lymphocytes is nutrient dependent but independent of Akt, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase, insulin growth factor-I, and serum. Cancer Res 2005;65(17):7800-8
- Majewski M, Korecka M, Kossev P, The immunosuppressive macrolide RAD inhibits growth of human Epstein-Barr virus-transformed B lymphocytes in vitro and in vivo: A potential approach to prevention and treatment of posttransplant lymphoproliferative disorders. Proc Natl Acad Sci USA 2000;97(8):4285-90
- Stallone G, Schena A, Infante B, Sirolimus for Kaposi's sarcoma in renal-transplant recipients. N Engl J Med 2005;352(13):1317-23
- Campistol JM, Gutierrez-Dalmau A, Torregrosa JV. Conversion to sirolimus: a successful treatment for posttransplantation Kaposi's sarcoma. Transplantation 2004;77(5):760-2
- Lebbe C, Euvrard S, Barrou B, Sirolimus conversion for patients with posttransplant Kaposi's sarcoma. Am J Transplant 2006;6(9):2164-8
- Boulanger E, Afonso PV, Yahiaoui Y, Human herpesvirus-8 (HHV-8)-associated primary effusion lymphoma in two renal transplant recipients receiving rapamycin. Am J Transplant 2008;8(3):707-10
- El-Salem M, Raghunath PN, Marzec M, Activation of mTORC1 signaling pathway in AIDS-related lymphomas. Am J Pathol 2009;175(2):817-24
- Roy D, Dittmer DP. PTEN on Chromosome 10 Is Phosphorylated in Primary Effusion Lymphoma and Kaposi's Sarcoma. Am J Pathol 2011;179(4):2108-19
- Aoki M, Blazek E, Vogt PK. A role of the kinase mTOR in cellular transformation induced by the oncoproteins P3k and Akt. Proc Natl Acad Sci USA 2001;98(1):136-41
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005;307(5712):1098-101
- Janes MR, Limon JJ, So L, Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med 2010;16(2):205-13
- Schenone S, Brullo C, Musumeci F, ATP-Competitive Inhibitors of mTOR: An Update. Curr Med Chem 2011;18(20):2995-3014
- Bhende PM, Park SI, Lim MS, The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious against follicular lymphoma. Leukemia 2010;24(10):1781-4
- Fan QW, Knight ZA, Goldenberg DD, A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006;9(5):341-9
- Chiarini F, Fala F, Tazzari PL, Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res 2009;69(8):3520-8
- Mazzoletti M, Bortolin F, Brunelli L, Combination of PI3K/mTOR Inhibitors: Antitumor Activity and Molecular Correlates. Cancer Res 2011;71(13):4573-84
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998;273(7):3963-6
- Fan QW, Cheng C, Hackett C, Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal 2010;3(147):ra81