Abstract
New therapies targeting critical elements of the cell cycle open novel and attractive avenues for the treatment of cancer patients. At present, the number of clinical trials that are registered with the European Organization for Research and Treatment of Cancer (EORTC) and with the US National Cancer Institute, which investigate the efficacy of Polo-like kinase 1 (Plk1) inhibitors against solid tumors and different types of leukemia is growing. Plks are important regulators of mitotic progression. Plk1, the best characterized mammalian Plk, has become an attractive target for cancer drug development, because most types of cancer appear to be addicted to the non-oncogene Plk1. Here, the authors discuss the role of Plk1 and the potential tumor suppressor gene Plk2 in acute myeloid leukemia (AML).
Acute myeloid leukemia (AML) is a rapidly developing neoplastic disease characterized by an accumulation of immature hematopoietic cells. Enhanced self-renewal and proliferation in combination with a differentiation block are important characteristics of the disease. Intensive, cytotoxic chemotherapy and allogeneic stem cell transplantation (SCT) are the current mainstays in the treatment of AML that allow curing the disease in a sizeable proportion particularly of younger patients Citation[1]. The vast majority of AML patients are elderly, however, and long-term outcome in these patients is dismal, as the efficacy and tolerability of intensive chemotherapy is poor and allogeneic SCT generally is not an option. In case of refractory or recurrent leukemia, results of salvage treatment are poor irrespective of age. This highlights the unmet clinical need for novel therapeutic approaches for AML that are more specific and thereby less toxic.
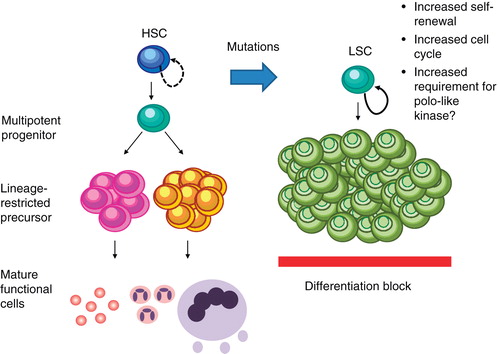
Recognition of the molecular heterogeneity of AML has led to the delineation of prognostically distinct AML subsets associated with therapeutically targetable genetic aberrations. Examples include the impressive responses observed with arsenic trioxide and all-trans retinoic acid (ATRA) in acute promyelocytic leukemia (APL) Citation[2] and the ability to target mutations in tyrosine kinases such as fms-like tyrosine kinase receptor-3 (FLT-3) by specific tyrosine kinase inhibitors Citation[3]. The clinical value of these novel compounds and their optimal combination with chemotherapy remain to be determined, and there have been unexpected setbacks; for example, combined treatment with the FLT-3 inhibitor sorafenib and chemotherapy failed to improve results achieved with chemotherapy alone in patients with unselected but also FLT-3 mutated AML Citation[4,5]. In addition, the small number of patients with leukemia of a particular, genetically defined subtype are a challenge for the clinical development of these novel treatment strategy. On the other hand, involvement of pathways such as the WNT pathway Citation[6,7], or alterations in the epigenetic regulation of genes do not seem to be restricted to certain AML subgroups and can be ameliorated by treatments with epigenetically active compounds such as the DNA methyltransferease (DNMT) inhibitors Citation[8]. As self-renewal and proliferation are among the hallmarks of AML development, cell cycle progression may also represent an attractive target ().
Figure 1. Plk1 in leukemic transformation. By increasing self-renewal and proliferation leukemic transformation may increase the requirement of Plk1.
Given the proven success of antimitotic therapies that change microtubule dynamics in the clinical treatment of cancer, it is reasonable to consider non-structural components of mitosis (e.g., cell cycle kinases like cyclin-dependent kinases, Aurora kinases A/B and Plks) as potential drug targets for therapy Citation[9]. The observation that loss-of-function alleles of the Drosophila homolog of Plk1, Polo, cause zygotic lethality indicated early on the critical role of Polo for cellular proliferation Citation[10,11]. To date, the human serine/threonine kinase Plk1 is the most comprehensively studied member of the Plk family (Plk1 – 5) Citation[12]. In eukaryotic cells from yeast to man, orthologs of Plk1 play essential roles during mitosis and meiosis Citation[10,11]. In vertebrate cells, Plk1 has been implicated in multiple steps of mitotic progression including the activation of Cdk1/cyclin B1, centrosome maturation, release of cohesion from chromosomes, activation of the anaphase-promoting complex/cyclosome (APC/C), formation of bipolar spindles and the regulation of the spindle assembly checkpoint (SAC) Citation[10,11]. Very intensive and fundamental research to understand the nature, the molecular functions and the regulations of polo-like kinases in several model systems is still continuing to this day.
Correlating with its multiple mitotic functions, Plk1 as marker of cellular proliferation is only detectable in adult tissues containing proliferating cells, for example, ovary, testis, spleen and epithelial tissues of the gastrointestinal tract Citation[13,14]. Remarkably, the levels of Plk1 in different types of tumors correlate inversely with patient survival, providing evidence for the role of Plk1 as prognostic marker Citation[15-19] and/or marker of metastatic disease Citation[20,21]. Recent genome-wide RNAi screens using various normal and isogenic cancer cells confirmed that cancer cells are addicted to a high level of non-oncogenic Plk1 for their viability Citation[22]. The recent analysis of an inducible RNAi model in transgenic mice supported the view that cancer cells and proliferating primary cells differ clearly in their dependency to Plk1 Citation[23,24]. With the development of antisense-oligonucleotide- and RNAi-based approaches loss-of-function screens were carried out with the aim of assessing the role of Plk1 in neoplastic phenotypes Citation[25-29]. The essential mitotic role of Plk1 in numerous types of cancer cells and its elevated expression in human cancers stimulated the validation of Plk1 as a novel drug target and the pharmaceutical development of many small molecule inhibitors Citation[12]. Targeting of Plk1 by novel compounds was judged to be specific based on a comparison with the biologic effects observed with RNAi. Based on the analysis of cancer cells lines different small molecule inhibitors were found to recapitulate the effects of Plk1 RNAi faithfully. BI 2536 is currently one of the most intensively studied Plk1 inhibitor and the first Plk1 inhibitor that entered the clinic Citation[30,31]. Both inhibitors, BI 2536 and BI 6727 Citation[4,32], completed Phase I studies and demonstrated good safety margins with preliminary evidence of antineoplastic activity representing the rational for further development in Phase II studies.
Investigations on the role of Plks in hematological diseases were scarce until recently, and documented only the elevated expression of Plk1 in lymphomas. Recent data describe the overexpression of Plk1 in hematological malignancies including AML compared with normal bone marrow mononuclear cells and normal peripheral blood mononuclear cells, suggesting that Plk1 is involved in leukemogenesis and serves as a promising target for the treatment of this disease Citation[33,34]. In contrast to normal hematopoietic stem cells, AML cell lines as well as primary human AML cells overexpress Plk1 and are sensitive to Plk1 inhibition in vitro. Remarkably, considering that the majority of the leukemic blasts are mainly blocked in G0/G1, the observed elevated levels of Plk1 expression in AML samples are surprising. Therefore, the role of Plk1 in proliferating and non-proliferating AML cells remains to be elucidated. Using Plk1-specific small interfering RNA (siRNA) or small molecule inhibitors like BI 2536 or GW843682X it could be demonstrated that leukemic cell lines depend on Plk1 for their proliferation and freshly isolated leukemic cells from patients including those that were refractory to conventional therapies depend on Plk1 for their clonogenic growth Citation[33,34]. The growth inhibition of leukemic cells by Plk1 inhibition was associated with G2/M arrest and the induction of apoptosis.
Considering the level of sequence identity of the kinase domain of Plk1 with those of Plk2, Plk3 and Plk4, it is conceivable that ATP-competitive inhibitors of Plk1 scarcely discriminate between Plk1 – 4. Still, Plk1 inhibitors that are currently in clinical trials like GSK 461364, NMS-P937, BI 2536 and BI 6727 show different selectivity profiles for the family members Plk2 and Plk3 Citation[12]. This is an important aspect also for the treatment of AML patients, because there are some indications that Plk2 and Plk3 can act as a tumor suppressors in vivo. For example, deletions in the long arm of chromosome 5, which are frequent chromosomal abnormalities in AML and human myelodysplastic syndrome (MDS), may contribute to the pathogenesis of these diseases by the loss of one or two tumor suppressor genes. Whether this chromosomal loss includes the Plk2 gene, which maps to chromosome 5 at 5q12.1–q13.2, has not been explored yet. To date, a tumor-suppressor function of Plk2 in hematopoietic diseases has been suggested in a study of B-cell malignancies, which describes a significant downregulation of Plk2 expression in a wide range of B-cell neoplasms due to a CpG methylation-dependent transcriptional silencing of Plk2 Citation[35]. Interestingly, the ectopic expression of Plk2 in Burkitt lymphoma cells induced apoptosis, suggesting a selective pressure to abolish the function of Plk2 in this type of cancer. The Plk2 CpG island was also found to be hypermethylated in 68.9 and 88.4% of AML and MDS cases, respectively Citation[36].
Further support for the role of Plk2 as a tumor suppressor came from a large-scale microRNA (miRNA) expression profiling study in core binding factor AML, an AML subtype representing 10 – 20% of primary AML Citation[37]. This study documents an overexpression of miR-126/126* that was associated with a partial demethylation of the CpG island in which miR-126/126* is located. Certainly, miR-126 has been shown to downregulate Plk2 levels and might thereby contribute to leukemogenesis. Thus, it remains to be elucidated whether ATP-competitive Plk1 inhibitors that also target Plk2 and Plk3 are optimal for cancer therapy due to their activity of inhibiting two potential tumor suppressor genes. Recent developments of small molecule inhibitors like poloxin, poloxipan, and purpurogallin. focus on Plk1 inhibition by targeting the less conserved polo-box binding domain (PBD) Citation[38-42]. Future studies need to answer the question whether Plk1 inhibition by targeting the PBD is the better cancer strategy to hit Plk1 specifically while sparing related Plk family members.
In the light of these considerations, a novel Plk inhibitor, NMS-937, which has an improved selectivity profile for Plk1 within the Plk family and is orally available, could have better long-term safety features in regard to effects in non-proliferating cells Citation[43]. The combination of a Plk inhibitor with cytarabine has evolved as a promising treatment strategy which prolonged survival in a xenograft model of AML Citation[43] and has proven safe in a Phase I trial of relapsed and refractory AML Citation[4,44].
Considering the critical role of both, Plks and Aurora kinases, in cell cycle regulation, their reported overexpression in hematologic malignancies including AML and their crosstalk, the rationale for exploring inhibitors of both of these kinase families as therapeutic strategies in AML is similar Citation[34,45]. Several inhibitors of the Aurora kinases are in fact in early-phase clinical testing Citation[46]. It is noteworthy that the majority of these compounds are multikinase inhibitors with activity not only against Aurora A and B but also against other kinases including ABL, FLT-3 and Janus kinase (JAK2). While these kinases play a role in hematologic malignancies, it is unclear whether the lack of selectivity typically observed with Aurora kinase inhibitors will enhance their potency or compromise their utility due to greater toxicity of multikinase inhibition. Mucositis and mucosal inflammation appear to be the major dose-limiting toxicities associated with Aurora kinase inhibitors Citation[47]. At present, the limited data that are available from the early stage clinical trials do not permit a comparison of Plk and Aurora kinase inhibitors. Nevertheless, it is conceivable that they may have variable clinical utility in different subtypes of leukemia, depending, for example, on the expression levels of Aurora kinases and Plks or the involvement of other kinases known to be involved in disease pathogenesis, for example, in leukemias with FLT-3 mutations.
Despite the attractiveness of Plk and Aurora kinase inhibitors as therapeutic agents for often highly proliferative diseases such as AML, it is almost certain that they eventually will need to be used in combination with other agents. Clinical development strategies for combination therapy will initially be restricted to drugs that are approved for treatment of AML and offer the potential for additive or synergistic activity. Accordingly, anthracyclines such as daunorubicin and cytosine arabinoside (ara-C) are the most plausible combination partners, even though the value of combining two agents with a cell cycle directed mode of action, that is, an Aurora kinase or Plk1 inhibitor with ara-C, may appear counterintuitive. However, preliminary data from preclinical studies with NMS-P937 and a Phase I trial exploring the administration of the Plk1 inhibitor BI 6727 with low-dose ara-C are promising and so far give no indication of antagonism between these two drugs Citation[4,43,44]. The potent myelosuppressive activity of Plk1 inhibitors makes them particularly appealing as a combination partner for induction chemotherapy, which typically is based on the combination of an anthracycline with ara-C. The tolerability of such combinations need to be determined in the setting of Phase Ib studies and will determine their feasibility during subsequent post-remission therapy.
The potential value of combining a Plk1 inhibitor with the topoisomerase inhibitor etoposide is suggested by an in vitro study demonstrating that in U937 AML cells, Plk1 participates in checkpoint recovery: inhibition of Plk1 by the GW843682X compound results in mitotic accumulation and apoptosis, and that when challenged with VP-16, inhibition of Plk1 prevented U937 cells from checkpoint exit Citation[48]. A theoretical caveat for combining these two classes of drugs is their shared propensity for inducing clinically relevant mucositis.
The concurrent use of a Plk1 inhibitor with a hypomethylating agent such as azacytidine is intriguing as it combines a cell cycle active compound with an agent considered to have antileukemic activity at the level of the leukemic stem cell, which is typically quiescent and should thus be largely spared by drugs acting primarily at the G2/M phase of the cell cycle. Clinical development of such a combination will need to focus particularly on the proper scheduling, as both drug classes have overlapping toxicities related to myelosuppression.
Expert opinion
In recent years, Plk1 has become one of the most attractive cancer targets in the field of antimitotics. Several Phase I/II studies using Plk1 inhibitors for monotherapy have demonstrated manageable, non-cumulative and predominately hematological side effects that reflect the inhibitor's predicted mode of action. First indications of anticancer activity are promising, but more efforts are required to improve treatment regimens, identify combinatorial agents with synergistic activity like cytarabine in AML models, and define biomarkers reflecting treatment response and patient characteristics that predict therapeutic success. Future clinical studies should include promising markers of Plk1 activity like serine 46-phosphorylation of the translational controlled tumor protein (TCTP) Citation[49] and an enzyme-linked immunosorbent assay (ELISA) Citation[50]. Since various lines of evidence support the correlation of p53 mutation in human cancer and cellular levels of Plk1, patient stratification will help to identify patients with high Plk1 expression, who may respond very well to therapeutical approaches aiming at Plk1 inhibition.
Declaration of interest
G Bug and OG Ottmann have each received an honorarium for participation at an advisory board meeting organized by Boehringer Ingelheim GmbH & Co. KG, Germany. No funding was received in preparation of this article. The authors have no other competing interests to declare.
Acknowledgements
T. Berg and K. Strebhardt received funding from the LOEWE-excellence cluster ‘Onkogene Signaltransduktion Frankfurt’ (OSF). Oliver G. Ottmann is an endowed Professor der German Jose Carreras Leukemnia Foundation. This work was supported by grants from the Deutsche Krebshilfe, the BANSS-Stiftung and Carls-Stiftung.
Notes
Bibliography
- Gupta V, Tallman MS, Weisdorf DJ. Allogeneic hematopoietic cell transplantation for adults with acute myeloid leukemia: myths, controversies, and unknowns. Blood 2011;117(8):2307-18
- Tallman MS, Andersen JW, Schiffer CA, All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med 1997;337(15):1021-8
- Small D. FLT3 mutations: biology and treatment. Hematology Am Soc Hematol Educ Program 2006;178-84
- Bug G, Schlenk RF, Muller-Tidow C, Phase I/II study of BI 6727 (volasertib), an intravenous polo-like kinase-1 (Plk1) inhibitor, in patients with acute myeloid leukemia (AML): results of the dose finding for BI 6727 in combination with low-dose cytarabine. Blood 2010;116(21):abstract 3316
- Serve H, Wagner R, Sauerland C, Sorafenib in combination with standard induction and consolidation therapy in elderly AML patients: results from a randomized, placebo-controlled phase II trial. Blood 2010;116(21):abstract 333
- Muller-Tidow C, Ji P, Diederichs S, The cyclin A1-CDK2 complex regulates DNA double-strand break repair. Mol Cell Biol 2004;24(20):8917-28
- Wang Y, Krivtsov AV, Sinha AU, The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010;327(5973):1650-3
- Claus R, Lubbert M. Epigenetic targets in hematopoietic malignancies. Oncogene 2003;22(42):6489-96
- Strebhardt K, Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat Rev Cancer 2008;8(6):473-80
- Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 2004;5(6):429-40
- Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol 2009;10(4):265-75
- Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010;9(8):643-60
- Holtrich U, Wolf G, Brauninger A, Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors. Proc Natl Acad Sci USA 1994;91(5):1736-40
- Yuan J, Horlin A, Hock B, Polo-like kinase, a novel marker for cellular proliferation. Am J Pathol 1997;150(4):1165-72
- Wolf G, Elez R, Doermer A, Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997;14(5):543-9
- Knecht R, Oberhauser C, Strebhardt K. PLK (polo-like kinase), a new prognostic marker for oropharyngeal carcinomas. Int J Cancer 2000;89(6):535-6
- Wolf G, Hildenbrand R, Schwar C, Polo-like kinase: a novel marker of proliferation: correlation with estrogen-receptor expression in human breast cancer. Pathol Res Pract 2000;196(11):753-9
- Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 2006;6(4):321-30
- Knecht R, Elez R, Oechler M, Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Research 1999;59(12):2794-7
- Strebhardt K, Kneisel L, Linhart C, Prognostic value of polo-like kinase expression in melanomas. JAMA 2000;283(4):479-80
- Kneisel L, Strebhardt K, Bernd A, Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J Cutan Pathol 2002;29(6):354-8
- Luo J, Emanuele MJ, Li D, A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the ras oncogene. Cell 2009;137(5):835-48
- Raab M, Kappel S, Kramer A, Toxicity modelling of Plk1-targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nat Commun 2011;2:395
- Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol 2006;26(6):2093-108
- Spankuch-Schmitt B, Bereiter-Hahn J, Kaufmann M, Strebhardt K. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J Natl Cancer Institute 2002;94(24):1863-77
- Spankuch-Schmitt B, Wolf G, Solbach C, Downregulation of human polo-like kinase activity by antisense oligonucleotides induces growth inhibition in cancer cells. Oncogene 2002;21(20):3162-71
- Liu X, Erikson RL. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci USA 2003;100(10):5789-94
- Spankuch B, Matthess Y, Knecht R, Cancer inhibition in nude mice after systemic application of U6 promoter-driven short hairpin RNAs against PLK1. J Natl Cancer Institute 2004;96(11):862-72
- Spankuch B, Heim S, Kurunci-Csacsko E, Down-regulation of Polo-like kinase 1 elevates drug sensitivity of breast cancer cells in vitro and in vivo. Cancer Res 2006;66(11):5836-46
- Steegmaier M, Hoffmann M, Baum A, BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Current Biol CB 2007;17(4):316-22
- Lenart P, Petronczki M, Steegmaier M, The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Current Biol CB 2007;17(4):304-15
- Rudolph D, Steegmaier M, Hoffmann M, BI 6727, a polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res 2009;15(9):3094-102
- Ikezoe T, Yang J, Nishioka C, A novel treatment strategy targeting polo-like kinase 1 in hematological malignancies. Leukemia 2009;23(9):1564-76
- Renner AG, Dos Santos C, Recher C, Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood 2009;114(3):659-62
- Syed N, Smith P, Sullivan A, Transcriptional silencing of Polo-like kinase 2 (SNK/PLK2) is a frequent event in B-cell malignancies. Blood 2006;107(1):250-6
- Benetatos L, Dasoula A, Hatzimichael E, Polo-like kinase 2 (SNK/PLK2) is a novel epigenetically regulated gene in acute myeloid leukemia and myelodysplastic syndromes: genetic and epigenetic interactions. Ann Hematol 2011;90(9):1037-45
- Li Z, Lu J, Sun M, Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA 2008;105(40):15535-40
- Yuan J, Sanhaji M, Kramer A, Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am J Pathol 2011;179(4):2091-9
- Lee KS, Song S, Erikson RL. The polo-box-dependent induction of ectopic septal structures by a mammalian polo kinase, plk, in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 1999;96(25):14360-5
- Reindl W, Yuan J, Kramer A, Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol 2008;15(5):459-66
- Reindl W, Yuan J, Kramer A, A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. ChemBioChem 2009;10(7):1145-8
- Watanabe N, Sekine T, Takagi M, Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J Biol Chem 2009;284(4):2344-53
- Valsasina B, Beria I, Alli C, NMS-P937, an orally available, specific, small molecule Polo-Like Kinase 1 inhibitor with antitumor activity in solid and haematological malignancies. Mol Cancer Ther 2012;11(4):1006-16
- Bug G, Muller-Tidow C, Schlenk RF, Phase I/II study of volasertib (BI 6727), an intravenous polo-like kinase (Plk) inhibitor, in patients with acute myeloid leukemia (AML): updated results of the dose finding phase i part for volasertib in combination with low-dose cytarabine (LD-Ara-C) and as monotherapy in relapsed/refractory AML. Blood 2011;118(21):abstract 1549
- Lucena-Araujo AR, de Oliveira FM, Leite-Cueva SD, High expression of AURKA and AURKB is associated with unfavorable cytogenetic abnormalities and high white blood cell count in patients with acute myeloid leukemia. Leuk Res 2011;35(2):260-4
- Boss DS, Beijnen JH, Schellens JH. Clinical experience with aurora kinase inhibitors: a review. Oncologist 2009;14(8):780-93
- Lowenberg B, Muus P, Ossenkoppele G, Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood 2011;118(23):6030-6
- Didier C, Cavelier C, Quaranta M, Evaluation of polo-like kinase 1 inhibition on the G2/M checkpoint in acute myelocytic leukaemia. Eur J Pharmacol 2008;591(1-3):102-5
- Cucchi U, Gianellini LM, De Ponti A, Phosphorylation of TCTP as a marker for polo-like kinase-1 activity in vivo. Anticancer Res 2010;30(12):4973-85
- Park JE, Li L, Park J, Direct quantification of polo-like kinase 1 activity in cells and tissues using a highly sensitive and specific ELISA assay. Proc Natl Acad Sci USA 2009;106(6):1725-30