
Abstract
Insulin-sensitizing thiazolidinediones (TZDs) correct a root cause of type 2 diabetes and potentially other diseases of metabolic dysfunction, including conditions ranging from oncologic, inflammatory, and neurodegenerative diseases. Importantly, compounds with this mode of action can modify disease progression, as opposed to simply mitigating symptoms. However, side effects have limited the use of marketed agents. Moreover, the same and additional issues have prevented development of newer agents, and no new compounds with this mode of action have been approved since 1999. Here we briefly review the drug discovery track record of compounds in the TZD class as well as several classes of compounds that have been designed with substitutes for the TZD ring, while maintaining and/or expanding the ability to directly activate peroxisome proliferator-activated receptor (PPAR) transcription factors. A key discovery that could change the course of drug discovery in this area is a newly identified mitochondrial target for the insulin sensitizers. This has allowed new drug discovery into molecules designed to maintain this mitochondrial interaction while specifically avoiding significant interactions with PPAR receptors. This commentary suggests that a fresh approach could pave the way for a new directed group of therapeutic agents with potential for disease modification of common metabolic disorders.
1. Insulin-sensitizing thiazolidinediones
The utility of a class of thiazolidinediones (TZDs) was an empirical finding pioneered by Sohda, Ikeda, Fujita, and colleagues at the Takeda company in the 1970s Citation[1]. These compounds came to be known as insulin sensitizers Citation[2] and although the Takeda analogs, ciglitazone followed by pioglitazone, were the first into clinical trials, troglitazone, an analog synthesized by Sankyo, was the first compound from this class (as Rezulin®), approved for the market in 1997. This event was followed by the approval of rosiglitazone (as Avandia®) and then pioglitazone (as Actos®) in 1999 Citation[3]. During the middle 1990s, some 10 years after the selection of the only 3 compounds with this action to be approved and marketed to treat type 2 diabetes, a strong case was made that these compounds exerted their activity by a direct activation of the nuclear transcription factor peroxisome proliferator-activated receptor γ (PPARγ). This conclusion was based primarily on the activity of the most potent activator of the original group of compounds, rosiglitazone, e.g., Citation[4]. The ability to find other compounds that activated PPARγ and which were also active as insulin sensitizers, particularly with respect to differentiation of adipose cells, led to the conclusion that insulin sensitizers were PPARγ agonists Citation[5]. Troglitazone was subsequently removed from the market because of a unique idiosyncratic hepatotoxicity. This event, together with the possibility to expand out of the chemical class, provided from the work done with rosiglitazone and related analogs that had demonstrated that a transcription factor could be used for screening (4), provided impetus for a number of drug-discovery programs to find new PPARγ agonists. There have been many other TZD molecules that have entered into development programs without success Citation[6,7]. The point of the current discussion is that the primary intent of later chemical programs was to increase potency, specifically, taking advantage of the developing insight into the predictive power of designing toward direct activation of PPARγ Citation[8]. The key structures of the important TZDs are shown in the left side of . It must again be pointed out that the eventually successful clinical candidates were all selected 10 years before the PPARγ hypothesis was developed.
Figure 1. Insulin sensitizer families. The family pedigrees of both TZDs (left panel) and non-TZDs are shown. Ciglitazone was the first insulin sensitizer tested in clinical trials. Troglitazone, rosiglitazone, and pioglitazone are the only insulin sensitizers ever to be approved for treatment of type 2 diabetes. Representative non-TZDs are shown on the right side of the page. Most of these compounds were modeled after Takeda's AD-5075, which was the most potent of the original Takeda analogs at differentiating 3T3L1 preadipocytes Citation[9], each with various substitutions for the TZD ring. None of these compounds were ever approved. Aleglitazar was the most recently discontinued.
![Figure 1. Insulin sensitizer families. The family pedigrees of both TZDs (left panel) and non-TZDs are shown. Ciglitazone was the first insulin sensitizer tested in clinical trials. Troglitazone, rosiglitazone, and pioglitazone are the only insulin sensitizers ever to be approved for treatment of type 2 diabetes. Representative non-TZDs are shown on the right side of the page. Most of these compounds were modeled after Takeda's AD-5075, which was the most potent of the original Takeda analogs at differentiating 3T3L1 preadipocytes Citation[9], each with various substitutions for the TZD ring. None of these compounds were ever approved. Aleglitazar was the most recently discontinued.](/cms/asset/6e3f478f-8380-4f91-9631-563cf1e8ccc3/ieid_a_839659_f0001_b.jpg)
2. Expansion into non-TZD chemical space
Given the observation that potency at activating PPARγ could provide a way for medicinal chemistry to expand evaluation of multiple chemical structures, many efforts were undertaken to expand the chemical space. However, as shown on the right side of , the starting point for many of the non-TZD clinical candidates that came from industry programs was a Takeda compound called AD-5075, which was disclosed as one of the most potent analogs from the original empirical workup of the class in terms of their ability to stimulate the differentiation of adipocytes Citation[9]. Potent PPARγ agonists were created by replacing the TZD ring with other acidic bioisosteres (the calculated pKa of the majority of the isoteres ranged between 6.4 for TZDs to 3.6 for alkoxy acids), with most of the efforts keeping the left-hand portion of the molecule fairly constant. We will not review all of these programs here, but some examples of various approaches are listed Citation[10-15]. Some of these changes resulted in the design and synthesis of analogs that also activated other nuclear receptors from the PPAR family. These have included compounds that affect some (γ, δ, or γ+α) or all (γ, α, δ) of the PPAR-related nuclear receptors Citation[16]. The one constant for both newer TZDs and the non-TZDs that have failed (summarized on the right side of ) is that all were selected with the idea of activating PPARγ. There have been no new market approvals of insulin-sensitizing agents since 1999, and all of these programs have now been stopped. There was a balanced activator of both PPARγ and PPARα, aleglitazar (Roche), which remained in clinical development until very recently Citation[17]. However, on July 10, 2013, Roche announced that this program had been discontinued because of “safety signals and lack of efficacy.” Judging from what was revealed in the corporate press release, the safety issues included some of the known PPAR effects and the lack of efficacy in this respect was due to the failure to show improvement in cardiovascular outcomes. Thus, the TZD clinical candidates that were eventually approved were selected before screening based on PPAR interactions were implemented and no clinical candidates have been approved for the treatment of diabetes since the PPAR-driven approach to drug discovery had been adopted ().
3. The case for a non-PPAR approach
Limited success of these endeavors might be due to the focus of all research activities on the wrong target(s) Citation[18] while a considerable portion of the insulin-sensitizing effect might involve anti-inflammatory or direct metabolic actions, including those in the mitochondrion Citation[19]. This concept will be expanded upon below. It should also be pointed out that recent evidence from long-term trials has suggested that of the two TZDs currently in clinical use, the weaker of the PPARγ activators, pioglitazone, has significant advantages over the stronger activator, rosiglitazone, in practice Citation[20-22]. These findings together with the failure of multiple efforts to successfully develop structurally diverse, potent PPAR activators for the treatment of diabetes support the view that an erosion of direct chemical activation of this transcription factor should be pursued Citation[18]. This hypothesis is being more directly tested with a series of TZDs that avoids binding to PPAR transcription factors; two of these ‘PPAR-sparing’ TZDs are in clinical development (see below). If this hypothesis is correct, insulin-sensitizing pharmacology as well as other pleiotropic effects of TZDs could result from defined direct action on non-PPAR targets and this should allow the insulin-sensitizing pharmacology unencumbered by PPAR-related side effects.
4. Direct mitochondrial effects of TZDs
A few years ago, Feinstein et al. reviewed the literature on the effects of TZDs on mitochondrial function Citation[23]. These authors pointed out that direct effects of these compounds on the mitochondria might include effects on ATP generation, AMP/ATP ratios, reactive nitrogen species, and reactive oxygen species, all of which could have important effects on cellular signaling mechanisms. In this respect, it is important to note that these compounds may have multiple, dose-dependent, effects on the mitochondria and that the relative effects of the compounds may differ with different analogs either due to intrinsic properties and/or cellular distribution. Feinstein reported that ciglitazone, pioglitazone, and troglitazone all rapidly increase glucose consumption and they also increased lactate production in astrocytes at concentrations not too different from circulating doses in vivo Citation[24]. Interestingly, Brunmair and colleagues have shown that a number of compounds of diverse structure that were actually designed to be either PPAR agonists or antagonists also have effects on the mitochondria that include inhibition of Complex I Citation[25,26]. There are also data demonstrating that TZDs, including troglitazone, pioglitazone, and rosiglitazone, can rapidly activate AMP kinase (AMPK) both in vitro and in vivo Citation[27,28] as well as in insulin-resistant animals Citation[29] wherein this mechanism appears to be involved in improvements in insulin sensitivity Citation[30]. While some of these effects may only occur at high doses and, in particular in vitro where there is no carrier protein to mitigate the free levels of drug available to the organelle, there may be more subtle effects specifically initiated at more pharmacologically relevant levels especially over time.
Effects on mitochondrial metabolism translate into different biological effects in different cell types and this will be considered in more detail below (Section 5). For example, Boyle et al. Citation[31], have shown that rosiglitazone stimulates the production of NO in endothelial cells through an AMPK-dependent mechanism. Such an effect may be related to the ability of rosiglitazone to lower blood pressure in vivo. There are also anti-inflammatory effects of the TZDs in endothelial cells that appear to involve both redox signaling Citation[32] and regulation of the activity of some MAP kinases Citation[33]. In other cells, activation of AMPK by this pathway would lead to important changes in lipid utilization or overall metabolism by mechanisms that are being well characterized Citation[34,35]. Activation of AMPK by these compounds could be secondary to a change in AMP/ATP ratios due to modulation of oxidative phosphorylation or secondary to other signaling (redox-related?) mechanisms (see below).
5. Mitochondrial binding and the discovery of mTOT, a mitochondrial target for TZDs
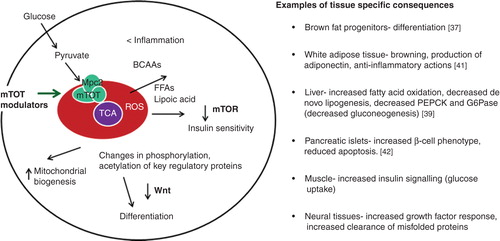
Direct effects on the mitochondria likely involve interaction with a specific protein or protein complex. Specific and saturable binding of 3H-pioglitazone to the mitochondria, competed by other active TZDs, has been demonstrated Citation[36]. A photoaffinity crosslinking approach identified a potential candidate as a small membrane protein that was named mitoNEET Citation[36]; however, the binding continued in the complete absence of mitoNEET Citation[37]. Importantly this binding demonstrates structural specificity such that close analogs devoid of cellular action also do not interact with this binding site. Further study established that the protein crosslinked by the drug analog photoprobe was in fact Mpc2, a component of the newly identified mitochondrial pyruvate carrier Citation[37], which resides in the internal mitochondrial membrane. This observation places the site of the interaction of these insulin sensitizers at the crossroads of metabolic regulation and identifies where to look for the “connection of the dots” that will help define how this mode of action operates at the molecular level. As shown in modification of mitochondrial target of thiazolidinediones (mTOT) results in alteration of nutrient-sensing pathways and modification of Wnt signaling Citation[37-42]. One might predict that these actions would produce a more widespread pharmacology applicable to many diseases with metabolic dysfunction, and this had in fact been predicted for insulin sensitizers, based on results in various animal models, even before the elucidation of these mechanisms Citation[3,18]. While it may be unclear whether there might be any longer-term issues with this pharmacology, at least we know that pioglitazone, which also impacts this mechanism, has been used clinically for over two decades and thus far with limitations that appear to be PPAR-related Citation[21,38].
Figure 2. Framework to view insulin-sensitizing pharmacology by modification of mTOT. The binding site for TZDs in the mitochondria is a complex in the inner mitochondrial membrane that has been called mTOT (mitochondrial target of thiazolidinediones). Key components of the complex are Mpc2, which is crosslinked by the drug analog crosslinker upon binding to intact membranes, and Mpc1, both of which are components of the pyruvate carrier. Regulation of the complex may determine the rate and delivery of pyruvate to help define the course of oxidative metabolism. The downstream effects that are known to occur upon the addition of the insulin sensitizers to various cell types include activation of AMPK, reduction of the activity of mTOR, and a metabolic break on Wnt signaling. These pathways are known to improve insulin signaling, but the consequences may vary in different cell types.
6. PPAR-sparing, mTOT-modulating compounds in development
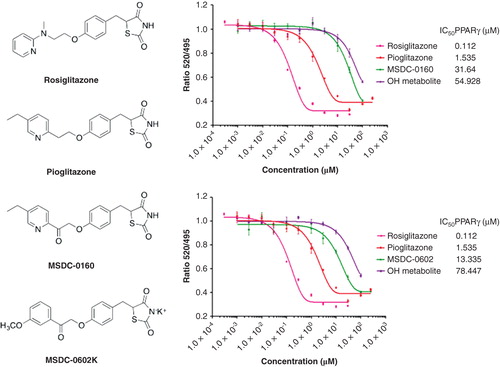
Two clinical candidates designed away from PPARγ activation and which maintain mTOT modulation are shown in . The first mTOT modulator to enter development was MSDC-0160. This compound and its major hydroxymetabolite have much reduced ability to bind to PPARγ (> 250- and > 50-fold, respectively, versus rosiglitazone and yet maintains the mTOT-modulating, insulin-sensitizing pharmacology Citation[37,38]). In a recently completed Phase IIb clinical trial in patients with type 2 diabetes, once daily treatment with the compound produced similar lowering of glucose (fasting plasma glucose and hemoglobin A1c) as did 45 mg pioglitazone, but with approximately half the reduction of hematocrit (less volume dilution) and half of the increase in high-molecular-weight adiponectin (produced from white fat stores). These data support the hypothesis that the ability to bind to and activate PPARγ can be removed from agents without compromising the ability of the compounds to be effective as antidiabetic agents Citation[38]. A second mTOT modulating compound, MSDC-0602, has recently completed a Phase IIa trial in diabetic subjects (NCT01280695) where it had effects similar to MSDC-0160. Studies in mice have demonstrated that this insulin-sensitizing pharmacology was independent of PPARγ. This separation was particularly clear in hepatocytes that were null for PPARγ expression Citation[39].
Figure 3. Relative effects of mTOT-modulating insulin sensitizers in development on binding to PPARγ. The structures of the two mTOT-modulating insulin sensitizers currently in clinical development are shown relative to rosiglitazone and pioglitazone. On the right side of the figure, the competition of the compounds for binding to human PPARγ is shown. For both MSDC-0160 (upper graph, right) and MSDC-0602 (lower graph, right), this activity is also shown for its major circulating reduced (OH) metabolite.
7. Expert opinion
This commentary suggests that a change in research focus is required to further the understanding of the complex pharmacology of “insulin sensitizers.” It is clear that compounds with this mode of action should no longer be viewed as simply direct activators/modulators of PPARγ. Obviously, there are major differences in the abilities of the TZDs to activate PPAR transcription factors and yet there is much overlap between the pharmacology of these agents. On the other hand, there is little doubt that activation of PPARγ can result in dose-limiting side effects such as sodium retention, edema, and potentially bone loss Citation[6,22]. The development of a new series of PPAR-sparing analogs, which are more specifically designed to be mTOT modulating, will be an important tool to utilize to elucidate the pathways responsible for improved insulin sensitivity and the pharmacology associated with these agents. Current evidence suggests that this pharmacology involves modification of the metabolism of nutrients by coordination of the mTOT complex, which includes the pyruvate carrier proteins. Activation of AMPK, inhibition of mTOR activation, and reduction of inflammatory kinase activities that produce a negative feedback on insulin signaling are all likely to be involved in the coordination of the pharmacological response. This likely involves modification of redox signals in response to nutrient utilization, which offers a rich possibility of biochemistry to consider as we proceed to understand the pharmacology in more specific terms Citation[40]. However, we must also consider that the overall response in vivo is likely to involve actions on multiple cell types (). Thus, a narrow consideration of the pharmacological impact in any single tissue will necessarily provide only a portion of the story. Given the complex pharmacology of these agents and their potential utility in multiple disease states, a more complete understanding of the biochemical pharmacology of these agents is likely to facilitate discovery and development of novel therapeutic agents as well as to have important implications for understanding metabolic disease. There should be an increased investigation of the specific direct effects of insulin sensitizers on mitochondrial function, particularly related to the newly identified complex in the internal membrane which houses the recognition site for the insulin sensitizers (mTOT). A fresh approach may provide renewed optimism in the search for new therapeutics to effectively treat and arrest costly metabolic disease.
Declaration ofinterests
J Colca is co-founder and part owner of Metabolic Solutions Development Company (MSDC). S Tanis is a stock holder of MSDC. W McDonald is an employee of and part owner of MSDC. R Kletzien is co-founder, part owner, and employee of MSDC.
Bibliography
- Sohda T, Meguro K, Kawamatsu Y. Studies on antidiabetic agents. IV. Synthesis and activity of the metabolites of 5-[4-(1-methylcyclohexylmethoxy)benzyl]-2,4-thiazolidinedione (ciglitazone). Chem Pharm Bull 1984;32:2267-78
- Hofmann CA, Colca JR. New oral thiazolidinedione antidiabetic agents act as insulin sensitizers. Diabetes Care 1992;15:1075-8
- Yki-Järvinen H. Thiazolidinediones. N Engl J Med 2004;351:1106-18
- Lehmann JM, Moore LB, Smith-Oliver TA, et.al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor [gamma] (PPAR [gamma]). J Biol Chem 1995;270:12953-6
- Olefsky JM, Saltiel AR. PPAR gamma and the treatment of insulin resistance. Trend Endocrinol. Metab 2000;11:362-8
- Peraza MA, Burdick AD, Marin HE, et al. The toxicology of ligands for peroxisome proliferator-activated receptors (PPAR). Toxicol Sci 2006;90:269-95
- Bloomgarden ZT. Treatment issues in Type 2 diabetes. Diabetes Care 2002;25:390-4
- Rucker C, Scaarsi M, Meringer M. 2D QSAR of PPARγ agonist binding and transactivation. Bioorg Med Chem 2006;14:5178-95
- Kletzien RF, Clarke SD, Ulrich RG. Enhancement of adipocyte differentiation by an insulin-sensitizing agent. Mol Pharmacol 1992;41:393
- Martin JA, Brooks DA, Prieto L, et al. 2-Alkoxydihydrocinnamates as PPAR agonists. Activity modulation by the incorporation of phenoxy substituents. Bioorg Med Chem Lett 2005;15(1):51-5
- Haigh SKB, Allen G, Birrell HC, et al. Non-thiazolidinedione antihyperglycemica agents. Part 3: the effects of stereochemistry on the potency of α-Methoxy-β-phenylpropanpic acids. Bioorg Med Chem 1999;7:821-30
- Azukizawa S, Kasai M, Takahashi K, et al. Synthesis and biological evaluation of (S)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acids: a novel series of PPAR gamma agonists. Chem Pharm Bull (Tokyo) 2008;56(3):335-45
- Liu KG, Lambert MH, Ayscue AH, et al. Synthesis and biological activity of L-tyrosine-based PPARgamma agonists with reduced molecular weight. Bioorg Med Chem Lett 2001;11(24):3111-13
- Lu Y, Guo Z, Guo Y, et al. Design, synthesis, and evaluation of 2-alkoxydihydrocinnamates as PPAR agonists. Biorgan Med Chem Lett 2006;16:915-919L
- Pirat C, Farce A, Lebegue N, et al. Targeting peroxisome proliferator-activated receptors (PPARs): development of modulators. J Med Chem 2012;55(9):4027-61
- Kliewer SA, Xu HE, Lambert MH, Willson TM. Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog. Horm Res 2001;56:239
- Dietz M, Mohr P, Kuhn B, et al. Comparative molecular profiling of the PPARα/γ activator aleglitazar: PPAR selectivity, activity and interaction with cofactors. Chem Med Chem 2012;7:1101-11
- Colca JR, Kletzien RF. What has prevented the expansion of insulin sensitisers? Expert Opin Investig Drugs 2006;15(3):205-10
- Colca JR. Insulin sensitizers may prevent metabolic inflammation. Biochem Pharmacol 2006;72(2):125-31
- Winkelmayer WC, Setoguchi S, Levin R, Solomon DK. Comparison of cardiovascular outcomes in elderly patients with diabetes who initiated rosiglitazone vs pioglitazone. Therapy Arch Intern Med 2008;168:2368-75
- Ryder REJ. Pioglitazone: an agent which reduces stroke, myocardial infarction and death and is also a key component of the modern paradigm for the optimum management of type 2 diabetes. Br. J. Diabetes Vasc Dis 2011;11:113-20
- Henry RR, Erikson D, Ciraldi TA. PPAR Agonists and the Future for Insulin Sensitizers. Br. J. Diabetes Vasc Dis 2012;12:206-10
- Feinstein DL, Spagnolo A, Akar C, et al. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol 2005;70(2):177-88
- Dello Russo C, Gavrilyuk V, Weinberg G, et al. Peroxisome proliferator-activated receptor gamma thiazolidinedione agonists increase glucose metabolism in astrocytes. J Biol Chem 2003;278:5828-36
- Brunmair B, Gras F, Neschen S, et al. Direct thiazolidinedione action on isolated rat skeletal muscle fuel handling is independent of peroxisome proliferator-activated receptor-{gamma}-mediated changes in gene expression. Diabetes 2001;50:2309-15
- Brunmair B, Staniek K, Gras F, et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes 2004;53:1052-9
- Konrad D, Rudich A, Bilan PJ, et al. Troglitazone causes acute mitochondrial membrane depolarisation and an AMPK-mediated increase in glucose phosphorylation in muscle cells. Diabetologia 2005;48:954-66
- LeBrasseur NK, Kelly M, Tsao T-S, et al. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab 2006;291:E175-81
- Lessard SJ, Chen Z-P, Watt MJ, et al. Chronic rosiglitazone treatment restores AMPK{alpha}2 activity in insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab 2006;290:E251-7
- Ye J-M, Dzamko N, Hoy AJ, et al. Rosiglitazone treatment enhances acute AMP-activated protein kinase-mediated muscle and adipose tissue glucose uptake in high-fat-fed rats. Diabetes 2006;55:2797-804
- Boyle JG, Logan PL, Ewar M-A, et al. Rosiglitazone stimulates nitric oxide synthesis in human aortic endothelial cells via AMP-activated protein kinase. J Biol Chem 2008;283:11210-17
- Ceolotton G, Gallo A, Papparella I, et al. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H Oxidase via AMPK-dependent mechanism arterioscler. Thromb. Vasc. Biol 2007;27:2627-33
- Lombardi A, Cantini G, Piscitelli E, et al.et al. new mechanism involving ERK contributes to rosiglitazone inhibition of tumor necrosis factor-{alpha} and interferon-{gamma} inflammatory effects in human endothelial cells. Arterioscler Thromb Vasc Biol 2008;28:718-24
- Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology 2006;21:48-60
- Ruderman NB, Park H, Kaushik VK, et al. AMPK as a metabolic switch in rat muscle, liver and adipose tissue after exercise. Acta Physiol Scand 2003;178(4):435-42
- Colca JR, McDonald WG, Waldon DJ, et al. Identification of a novel mitochondrial protein (‘mitoNEET’) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab 2004;286:252-60
- Colca JR, McDonald WG, Cavey GS, et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)-relationship to newly identified mitochondrial pyruvate carrier proteins. PLOS ONE 2013;8:e61551-1-10
- Colca JR, VanderLugt JT, Adams WJ, et al. Clinical proof of concept with MSDC-0160, a prototype mTOT modulating insulin sensitizer. Clin Pharmaco Thera 2013;93:352-9
- Chen Z, Vigueira PA, Chambers KT, et al. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor -sparing thiazolidinedione. J Biol Chem 2012;287:23537-48
- Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxidant and Redox Signaling 2012;16:1323-67
- McDonald WG, Cole SL, Holewa DD, et al. New insulin sensitizers produce differentiation of brown-like adipose cells from a subcutaneous fat depot and increase secretion of adiponectin in vitro. Diabetologia 2011;54(Suppl 1):S15
- Rohatgi N, Aly H, Marshall CA, et al. Novel insulin sensitizer modulates nutrient sensitizing pathways and maintains β-cell phenotype in human islets. PLoS ONE 2013;8:e62012-1-13