Abstract
Protein kinase inhibitors have proved to be effective and well tolerated in special form of malignant diseases in which targeted kinases play a central role in the development and progression of the malignant clone. In addition, the development of acquired drug resistance, due to new mutations or clonal evolution, during treatment is common. Methods for measuring the activity and predicting the efficacy of such compounds are warranted for evaluating individual responses to treatment, particularly in a context of widespread preclinical and clinical studies of protein kinase inhibitors in patients with heterogeneous myeloid malignancies.
In this issue of Expert Opinion on Investigational Drugs, Fan et al. are highlighting the importance of analyzing drug activity to individualize therapy, an emerging approach of highly interest in the treatment of cancer patients Citation[1]. This is supported by evidence that drug can induce different effects among some patients and, on the other hand, induce only opposite unwanted effect in other patients Citation[2,3]. Fan et al. describe a novel nanoscalar proteomic assay to identify the susceptibility of myelodysplastic syndrome (MDS) patients to rigosertib, a dual non-ATP inhibitor of polo-like kinase 1 (PLK1) and phosphoinositide 3-kinase pathways (PI3K) Citation[1].
A protein kinase modifies other proteins by chemically adding phosphate groups to them Citation[4]. The human genome contains about 500 protein kinase genes, and they constitute about 2% of all human genes. They can be further divided in tyrosine kinases and serine-threonine kinases, depending on their ability to phosphorylate different amino acid residues; or they can be divided as receptor protein kinases and nonreceptor protein kinases, depending on their cellular location. Some of the most important protein kinases of interest of pharmacological targeting are highlighted in and . The receptor protein kinases are membrane-spanning cell surface proteins that play critical roles in the transduction of extracellular signals to the cytoplasm. Nonreceptor protein kinases, on the other hand, relay intracellular signals.
Table 1. Protein kinases can be divided as receptor protein kinases and nonreceptor protein kinases, depending on their cellular location.
Figure 1. Central protein kinases to be target for treatment of malignant diseases. A wide variety of cytokines and growth factor can bind membrane-bound receptor tyrosine kinases and activate intracellular cascades involving several intracellular protein kinases, representing them as potential target for new therapeutic agents.
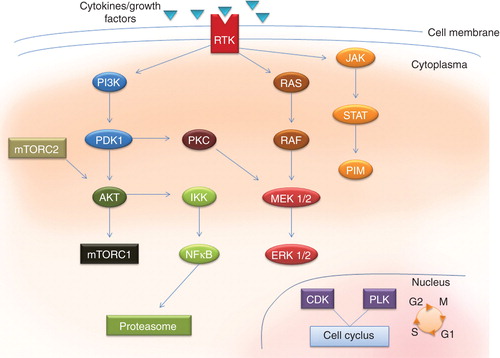
Human PLK1 is essential during mitosis and in the maintenance of genomic stability. PLK1 is over expressed in human tumors and has prognostic impact in cancer, indicating its involvement in carcinogenesis and its potential as a therapeutic target Citation[5]. The use of different PLK1 inhibitors has increased our knowledge of mitotic regulation and allowed us to assess their ability to suppress tumor growth in vivo. Many ATP-competitive inhibitors are heterocyclic systems able to interact with the unique features of the PLK1-binding site. Other inhibitors target regions outside the ATP pocket, such as the substrate binding domain or a hydrophobic pocket, formed when the kinase is in the inactive conformation Citation[5]. Moreover, a combination of polo-like kinases inhibitors with other anticancer drugs might offer new opportunities for cancer therapy Citation[5].
The PI3K/mammalian target of rapamycin/AKT (PI3K/mTOR/AKT) signaling pathway is central to the promotion of cellular growth, proliferation and survival, highlighting proteins within the PI3K/mTOR/AKT pathway as attractive targets for therapeutic intervention in cancer Citation[6,7]. PI3K/AKT activation may result from genetic hits affecting different components of the pathway Citation[6]. The connections between the PI3K and mTOR are multiple and complex, including common substrates, negative feedback loops or direct activation mechanisms Citation[6]. There is an emerging interest for targeting this pathway in malignant diseases, and newer and more potent inhibitors which may overcome resistance problem are emerging Citation[1,6,8].
Targeted therapy with tyrosine kinase inhibitors (TKIs) has been a story of success in chronic myeloid leukemia (CML), since the initial introduction and approval of imatinib over a decade ago Citation[4]. Kinase inhibition has continued to be an area of active research and development in the care of patients with hematological malignancies Citation[4]. This is currently emphasized by the introduction of janus kinase (JAK) inhibitors in treatment of another group of myeloid malignancies, namely the Philadelphia chromosome negative myeloprolifertive neoplasm (MPN); polycytemia vera (PV), essential thrombocythemia (ET) and myelofibrosis (MF) Citation[9]. However in field of the two other major group of myeloid malignancies, acute myeloid leukemia (AML) and the preleukemic MDS, the successes story from CML has not been followed, and there are still several obstacles to be solved before targeted therapy can be an option also among these patients.
In the cases of AML and MDS most expectations so far have been raised to inhibitors of the FMS-like tyrosine kinase 3 (FLT3), which is mutated in roughly 30% of AML patients and to a less extent in MDS Citation[10,11]. Constitutively active, mutant FLT3 leads to complex protein interactions inducing a cascade of protein phosphorylation events in downstream targets including MAP kinase, signal transducer and activator (STAT) and PI3K/AKT pathway Citation[11]. This facilitates proliferation and survival of leukemic blasts. On a theoretical basis, inhibition of FLT3 represents an ideal target in AML and MDS, and several FLT3 inhibitors have been developed and entered clinical trials. Unfortunately, their clinical effects so far seem limited, due to the engagement of mutational resistance mechanisms and probably only a minority of patients will benefit from them. Similar outcome was seen for several other protein kinase inhibitors under clinical trials for AML and MDS.
Why are TKIs so effective in CML and JAK inhibitors seeming promising in MPN, while targeted therapy so far have been rather disappointing in MDS and AML? First, in general AML and high risk MDS are far more aggressive diseases, illustrated by the fact that majority of AML patients untreated will die within months. It is unlikely that maintenance therapy with a single agent, like for CML, will keep the disease in a steady state.
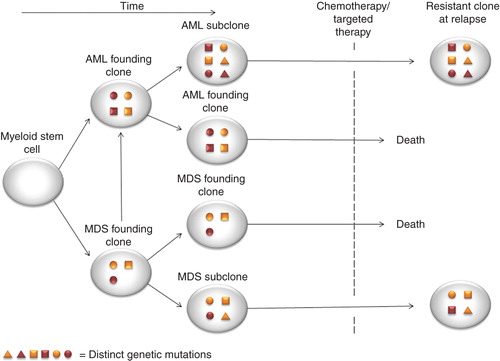
Second, branching clonal evolution seems to occur in both MDS, AML and in the transforming of MDS to AML, with different mutations existing in founding clones versus subclones Citation[12,13]. In other words, targeted therapy against the founding clone harboring a driver mutation, can select growth enhancement for more resistant and aggressive subclones which will spread at relapse (). Noteworthy, it is actually not clear whether treatments induce new mutations, only select therapy-insensitive subclones or if it is leukemia initiating cells persisting following therapy, which eventually give rise to the refractory clones at relapse.
Figure 2. Model of AML and MDS mutational and clonal evolution and mechanism of resistance to therapy. Mutations with myeloid stem cell or myeloid progenitor cell occur and develop to AML or MDS phenotype is followed. Branching evolution occurs as progression mutation occurs and gives rise to subclones. These subclones can be resistance to chemotherapy or targeted therapy obtained, and at relapse the original subclone with mutation predicting for resistant to therapy will occur both in AML and MDS.
Third, MDS/AML are heterogeneous diseases at a cytogenetic and molecular scale, which is illustrated by the diversity of their phenotypes () Citation[11,14]. This sharply contrasts with CML, in which the Philadelphia chromosome t(9; 22) translocation, by giving rise to the oncoprotein BCR-ABL, is self-sufficient to generate the disease. Hence, BCR-ABL TKIs such as imatinib dramatically modified CML natural evolution and outcome Citation[4]. This is also the case of MPN, there the common JAK2V617F mutation, which is seen in 95% of patients with PV and 50 – 60% of patients with ET and MF. Hence both CML and a majority of Philadelphia chromosome negative MPN, i.e., PV, ET and MF, are clearly driven by tyrosin kinases, either as a fusion oncoprotein; BCR-ABL, or an activating mutation; JA2V617F. Therefore BCR-ABL TKIs such as imatinib dramatically modified CML natural evolution and outcome Citation[4], and JAK TKIs have showing effect in relieving splenomegaly and hepatomegaly and constitutional symptoms in addition to modulating inflammatory cytokines in MF Citation[9]. This is highly in contrast to AML and MDS, there a diverse variety of cytogenetic alterations and molecular genetic mutations are found, including oncogenic driving kinases, but also a series of other abnormalities involving a variety of cellular process including transcription factors, DNA-methylation genes and other signaling genes () Citation[10,14]. Hence in most MDS and AML cases there is at least two and often more mutation driving the leukemic process Citation[10], and hence target therapy with TKIs or other agents will consequently be less effective Citation[11]. An important exception in this setting is the AML subtype FAB M3, the acute promyelocytic leukemia (APL) variant, there a single translocation, namely the t(15;17), is believed to be the main driver for the leukemic phenotype () Citation[10,13]. This translocation merge the promyeolocytic leukemia (PML) protein with the retinoic acid receptor alpha (RARA), giving origin to the oncoprotein PML-RARA, leading to blocking in differentiation and maturation of myeloid precursor cells Citation[13]. Although PML-RARA is not a kinase, this AML subtype is an example of a strategy where targeted therapy successfully have lead to increase response rates and improving overall survival. Pharmacological targeting with all-trans retinoic acid (ATRA) or arsenic trioxide (As2O3) leads to high response rates, making this subtype of AML highly curable. This illustrate that oncogenic addiction probable exist also in MDS and AML if the target of treatment is the driver mutation, making targeted therapy with one, or most probably, several agents still an option Citation[1,11].
Table 2. The main morphological, clinical, cytogenetical, molecular genetics and treatment options in the major myeloid malignancies.
Fourth, genetic mutational resistance may occur under TKI treatment. This phenomenon is also known from TKI treatment in CML where new mutations, especially the T315I gatekeeper mutation, lead to resistance against conventional BCR-ABL TKIs and consequently disease progression Citation[4]. New drugs designed to overcome such mutational resistance are currently entering clinical trials Citation[4]. Such new mutations within the tyrosine kinase domain in FLT3 are also likely to occur during TKI treatment in AML; however, the clinical consequences are sharply different in CML compared to MDS/AML, with a progressive and rapid disease evolution following relapse in the latter case Citation[15].
Finally, protein kinase inhibitor could also have unwanted and unexpected effects. Indeed, positive feedback signal may be induced following the specific inhibition of a given signaling pathway to maintain a balance between negative and positive inputs on proliferation and survival, which should prompt us to investigate new therapies which are synthetically lethal with one given kinase inhibitor Citation[2,4,8]. The nonspecific targets, i.e., wild-type protein just like JAK2 normal protein and JAK2V617F protein in classical MPD Citation[9], or out-of-target just like the inhibition of the tyrosine kinase family SRC with TKIs in CML, probably also explain side effects seen with TKI treatment. The later explaining at least in part the secondary side-effects of dasatinib and why a significant portion of patients have to withdrawal from medication Citation[4].
Taken these facts into considerations, is it likely that AML/MDS will benefit for any protein kinase inhibitors? With the exception of APL, it seems simplistic to hypothesize that a single agent will eliminate the malignant clone in MDS or AML and definitively restore normal hematopoiesis Citation[11]. The specter and scale of protein kinases, often named the kinome, is heterogeneous and overlapping (, ), and activation of redundant and alternative signaling pathways seem likely to occur Citation[4]. In this setting, the combination of different new and specific kinase inhibitors with conventional chemotherapy or new monoclonal antibodies could represent a more plausible strategy Citation[11]. Identification of driver and passenger molecular mutations and appropriate use of targeted therapies, either alone or in combinations, may eventually revolutionize the treatment of MDS/AML.
In this setting, the approach to detect early in the clinical setting the particular sensitivity for each patient to protein kinase inhibitors is therefore crucial Citation[1]. Nanoscale proteomic technology can be used to uncover unanticipated changes in protein signaling, and hence therapy response in vivo Citation[1]. Such approaches are of high interest to evaluate subtle shifts in protein abundance and modification that maybe useful for the evaluation and discovery of new drugs in cancer therapy Citation[1]. Taken together, this indicate a future and exploring field of drug discovery and drug evaluation in the time to come in the search for target therapy in heterogeneous malignant diseases.
Expert opinion
Myeloid malignancies are characterized by high level of heterogeneity with regard to clinical, morphological, cytogenetical and molecular genetical features. New targeted therapy is warranted and a high variance among responders and nonresponders to new therapeutic advances should be expected. Early detecting of intracellular changes among treated patient have the potential to predict which patient who will respond and who will not. Such approaches are of high interest in the setting of cancer in general and myeloid malignancies in special, and should be further explored. Rigosertib is one of the new drugs entering clinical trials of AML and MDS, and in this context the article by Fan et al. regarding early detection of responders and non-responders is of high interest Citation[1].
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Notes
Bibliography
- Fan AC, O'Rourke JJ, Dave R, Felsher DW. Real-time nanoscale proteomic analysis of the novel multi-kinas pathway inhibitor rigosertib to measure the response to treatment of cancer. Expert Opin Investig Drugs 2013;22(11):1495-509
- Sawyers CL. Will kinase inhibitors have a dark side? N Engl J Med 2006;355(3):313-15
- Reikvam H, Tamburini J, Skrede S, et al. Antileukemic effect of PI3K-mTOR inhibitors in acute myeloid leukemia- gene expression profiles reveal CDC25B expression as determinate of pharmacological effect. Br J Haematol 2013, In press
- Sawyer TK, Wu JC, Sawyer JR, English JM. Protein kinase inhibitors: breakthrough medicines and the next generation. Expert Opin Investig Drugs 2013;22(6):675-8
- Tsykunova G, Reikvam H, Ahmed AB, et al. Targeting of polo-like kinases and their cross talk with Aurora kinases–possible therapeutic strategies in human acute myeloid leukemia? Expert Opin Investig Drugs 2012;21(5):587-603
- Willems L, Tamburini J, Chapuis N, et al. PI3K and mTOR signaling pathways in cancer: new data on targeted therapies. Curr Oncol Rep 2012;14(2):129-38
- Reikvam H, Nepstad I, Bruserud Ø, Hatfield K. Pharmacologic targeting of the PI3K/mTOR pathway controls release of angioregulators from primary human acute myeloid leukemia cells and their neighboring stromal cells. Oncotarget 2013;4(6):830-43
- Naing A. Overcoming resistance to mTOR inhibition for enhanced strategies in clinical trials. Expert Opin Investig Drugs 2013;22(6):679-85
- Tam CS, Verstovsek S. Investigational Janus kinase inhibitors. Expert Opin Investig Drugs 2013;22(6):687-99
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368(22):2059-74
- Ferrara F. New agents for acute myeloid leukemia: is it time for targeted therapies? Expert Opin Investig Drugs 2012;21(2):179-89
- Walter MJ, Shen D, Ding L, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 2012;366(12):1090-8
- Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012;150(2):264-78
- Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011;364(26):2496-506
- Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012;485(7397):260-3