
ABSTRACT
On March 6th 2015, the Food and Drug Administration (FDA) approved filgrastim-sndz (Zarxio) as the first biosimilar in the United States (US) for all indications of the reference product. Filgrastim-sndz is a biosimilar of Amgen’s Neupogen and is mainly used to treat neutropenia in cancer patients receiving chemotherapy. This article presents a summary of the analytical and clinical studies submitted by Sandoz and describes how the information was integrated to provide the ‘totality of the evidence’ leading to the approval of the biosimilar.
1. The totality of evidence concept
The totality of evidence includes the foundational comparative analytical and functional characterization studies, and the supportive nonclinical and clinical studies ( and ).[Citation1–Citation4] Despite their complexity, state-of-the-art analytical technologies allow a thorough physico-chemical characterization of biologics today. Combined with a detailed functional characterization and increasing understanding of the structure–function relationships, these studies already allow a meaningful prediction of the biological function and clinical performance. Subsequent nonclinical and clinical studies are needed to address any residual uncertainty about biosimilarity, the extent of which is defined by the confidence in the analytical similarity.
Figure 1. Totality of the evidence. (a) The totality of the evidence concept. The analytical characterization is the foundation of the step-wise approach to achieve biosimilarity. (b) Summary of the analytical and clinical studies performed using the US-licensed reference and the EU-approved comparator products. (c) Comparison of the tier 1 attribute ‘bioactivity’ between filgrastim-sndz, the US-licensed and the EU-approved reference product. Statistical evaluation was performed by equivalence testing of the means. Equivalency margin ± 1.5σC and 90% confidence interval. (d) Equivalent absolute neutrophil count (ANC) responses across all doses and routes of administration between the biosimilar and the US-licensed and the EU-approved reference product.
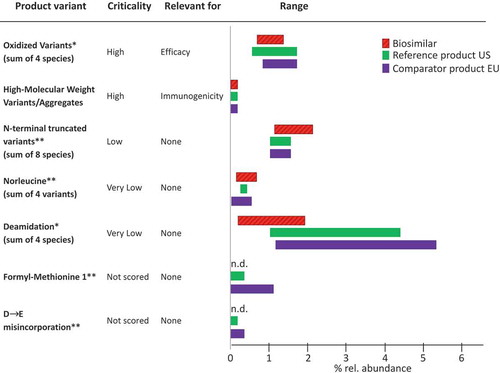
Figure 2. Comparison of the micro-heterogeneity of the biosimilar and the US-licensed reference and the EU-approved comparator product. Product variants of 17 lots of filgrastim-sndz and >50 lots of the reference/comparator product were analyzed by reversed-phase chromatography (RP-HPLC) and size exclusion chromatography (SEC), respectively. n.d.: not detected; *stability indicating, thus dependent on age of lot; **measured by mass spectrometry.
2. Analytical characterization – assessing the residual uncertainty
After a full characterization of the reference product and understanding the variability and consistency of the reference product attributes, the first step toward achieving biosimilarity is the comparative analytical characterization to demonstrate ‘high similarity’ between the biosimilar and the reference product. In the case of the biosimilar filgrastim-sndz (trade name Zarxio), we used multiple state-of-the-art analytical technologies to perform an in-depth characterization of the physico-chemical as well as the functional properties of the product. Filgrastim is a well-researched, relatively small (18 kDa) non-glycosylated protein expressed in E. coli bacteria, yet even in such a ‘simple’ biologic we identified a substantial number of product attributes (referred to by regulators as quality attributes) which were evaluated throughout development and assessment of biosimilarity (). The quality attributes were ranked according to their impact on the clinical parameter efficacy, safety and immunogenicity, based on knowledge from the literature, from functional characterization and from clinical experience gained in clinical studies and clinical practice.
Table 1. Criticality assessment of product quality attributes.
This so-called criticality assessment of quality attributes is continuously enhanced and complemented as development progresses from the initial phase, in which the focus is on achieving high similarity via targeted process development, all the way to commercial manufacturing, during which product consistency is the primary objective. Quality attributes with the greatest impact on potential clinical outcome are defined as critical quality attributes, and they must be kept within appropriate limits to ensure the desired product quality. During their review and evaluation the Food and Drug Administration (FDA) uses the criticality assessment as the basis to determine which quality attributes are most relevant to the determination of biosimilarity (tiered approach). Specifically attributes which are involved in the mechanism(s) of action for each indication for which approval is sought receive the most attention (tier 1 attributes).[Citation3,Citation4] In the assessment of filgrastim-sndz, potency (biological activity) and protein concentration (content) were categorized as tier 1 attributes.[Citation5,Citation6] For these, equivalence of the means of the biosimilar and the reference product was assessed using a two one-sided t-test (TOST). Filgrastim-sndz meets the equivalence criteria in comparison to both, the reference product from the US and the comparator product from the EU, which constitutes an important contribution to the totality of the evidence (, content not shown).
On top of the tier 1 attributes, a number of additional quality attributes were assayed and evaluated (). The identical amino acid sequence was confirmed using state-of-the-art technologies including high-resolution mass spectrometry. Correct folding (higher-order structure) and target-binding were assayed using 2D-NMR spectroscopy, surface plasmon resonance and other methods (). Again, the biosimilar, the reference and the comparator product were found to be highly similar. As a consequence of being manufactured using living organisms, all biologics exhibit some batch-to-batch variability in their quality attributes, which is mainly reflected in what is referred to as microheterogeneity of product variants ().[Citation7] Consequently, a thorough investigation of the product’s microheterogeneity is another important piece of the totality of the evidence. Both number and abundance of such product variants in the biosimilar were found to be highly similar to the reference and comparator product and generally of low abundance. In addition, process-related impurities such as host cell proteins were found to be present in similar, and very low, amounts, and finally, the products showed highly similar stability profiles.[Citation5] The high similarity to the comparator product approved in the EU and the equivalence between the US-licensed reference and the EU-approved comparator product is of key importance because it established analytical and functional bridges between the products to support the relevance of data generated from clinical and nonclinical studies using the EU-approved comparator product ().
3. Nonclinical and clinical studies – resolving the residual uncertainty
Nonclinical and clinical studies are used to confirm similarity and resolve residual uncertainty about biosimilarity that may have emerged with the analytical evaluation. The goal is to document that there are no clinically meaningful difference between the biological product and the reference product in terms of the purity, and potency. Additionally, nonclinical and clinical studies are valuable measures to confirm no meaningful difference in terms of safety, for example, in the incidence of anti-drug-antibody formation. However, it is conceptually important to understand that a sponsor cannot test a product into biosimilarity using clinical studies because clinical studies are often the least sensitive in detecting differences between two biological products. Consequently, the preceding demonstration of ‘high similarity’ in the comparative analytical and functional characterization is indispensable in the step-wise approach of achieving biosimilarity and also a statutory requirement. In addition, high analytical and functional similarity is the foundation for extrapolation of indications as only two highly similar molecules can be expected to act in the same way in all patient populations.
Sandoz performed an extensive program of nonclinical and clinical studies for the biosimilar filgrastim-sndz (), building on the supportive comprehensive data set which formed the basis of the approval by the European Medicines Agency (EMA) in 2009.[Citation8] Two pivotal studies were performed specifically for the US and were performed head-to-head against the US-licensed reference product: one study evaluating pharmacokinetics and pharmacodynamics () in healthy volunteers and one study evaluating safety and efficacy in breast cancer patients undergoing chemotherapy. [Citation9]
Table 2. PK/PD and clinical studies of filgrastim-sndz.
The EU-approved comparator product was used in a total of five animal studies comparing pharmacodynamics, toxicity, toxicokinetics, and local tolerance and in five PK/PD studies in healthy subjects.[Citation5,Citation6] Since the absolute neutrophil count (ANC) is a well-established PD marker that drives diagnosis and treatment in patients and donors and which can be sensitively measured in healthy volunteers, the EMA approval was based on establishing PK bioequivalence and PD equivalence across a wide range of doses (1–10 mcg/kg) using single and multiple s.c. or i.v. applications.[Citation8] An additional single-arm patient study to rule out any unexpected safety or immunogenicity concerns was sufficient to obtain marketing authorization in Europe in 2008.
The additional PK/PD study [Citation10] had the primary purpose of establishing PK/PD equivalence against the US-licensed reference product to satisfy the corresponding statutory requirement. The comparison of the PK profiles showed bioequivalent, but slightly lower exposure following administration of the biosimilar as compared to the reference product. These differences can be explained by the fact that for filgrastim-sndz a different formulation had to be used during development due to IP restrictions (the biosimilar uses a glutamate buffer at ph 4.4 vs. the reference product that uses an acetate buffer at pH 4.0). An exploratory study confirmed identical PK properties for filgrastim-sndz when formulated in the reference products’ buffer, indicating that the slightly lower numerical exposure is due to the difference in the buffer and not due to differences in the molecule and the FDA and Oncology Drugs Advisory Committee did not consider this slight difference clinically relevant.[Citation5,Citation6] The healthy volunteer studies established equivalent ANC responses across all doses and routes of administration () and also confirmed superimposable response profiles for the CD34+ cell counts, which is the relevant marker for the mobilization indications.[Citation5,Citation6]
For the biosimilar filgrastim-sndz, the FDA concluded that the clinical and commercial product is ‘highly similar’ to the US-licensed reference product and that the analytical similarity data does not raise residual uncertainties.[Citation5,Citation6] So for filgrastim-sndz, the clinical program served as a confirmation of biosimilarity rather than to address uncertainties coming from the analytical data. The additional confirmatory clinical trial in breast cancer patients was designed to investigate the effects of multiple switches to address interchangeability as part of further interactions with the FDA as the standards for interchangeability are still being developed.[Citation9]
Based on an extensive analytical bridge established between the biosimilar and both the US-licensed reference and the EU-approved comparator product, data generated across all studies conducted was considered relevant for FDA approval. Post-marketing pharmacovigilance is an important additional measure to provide evidence that a biosimilar is comparable to the reference product in terms of its safety profile. For the biosimilar filgrastim-sndz, the FDA also considered the extensive post-marketing experience gained based on approval in more than 60 countries and more than 7.5 million patient-days of exposure. Importantly, across all studies as well as based on the post-marketing experience, filgrastim-sndz showed a similar safety profile as compared to the reference and the comparator product and no signs of immunogenicity were observed further substantiating the similarity to the reference product with respect to safety, purity and potency.
4. Conclusion
The biosimilar filgrastim-sndz is an example of a thorough program used to establish biosimilarity – and the first in the US. The biosimilar and the reference product are highly similar in all aspects and there are no clinically relevant differences. The few minor differences, such as the formulation buffer, were shown to not be clinically meaningful. Importantly, extrapolation of indications as defined in the FDA’s guidelines was granted for the biosimilar. The data demonstrating high similarity and the fact that filgrastim exerts all of its pharmacological effects in the five different indications through the G-CSF receptor allowed the extrapolation of use of the biosimilar to all indications of the reference product although only one indication was clinically studied.
5. Expert opinion
A biosimilar is systematically designed to be as close to the reference product as the reference product is to itself considering variability observed from batch-to-batch and manufacturing changes over time. In fact the EMA consideres an approved biosimilar to be essentially the same active substance.[Citation11] This is also the scientific basis for the extrapolation of indications as a key understanding of proteins is that structure drives function. Consequently, the active substance of the biosimilar as well as the active substance of the reference product will bind to the same target(s)/receptor(s) with the same affinity and will therefore induce the same signaling cascades which ultimately results in the same clinical effects. The notion that the ‘process is the drug’ is a common misunderstanding with regard to biologics as demonstrated with the approval of major manufacturing changes by regulatory authorities using comparability exercises.[Citation12] The continuum of comparability extends to biosimilarity, and thus, the goal of the subsequent clinical studies is therefore to confirm biosimilarity instead of reestablishing safety and efficacy which have been determined already by the reference product. As with any new drug on the market, post-marketing studies will additionally confirm a highly similar clinical profile for the biosimilar compared to the reference product. The tailored clinical development program as well as the extrapolation of indications also support access to affordable high-quality biologics as already demonstrated in the EU.[Citation13] The ‘totality of the evidence’ is a robust, science-based concept which provides an overall assessment of biosimilarity from multiple comparative evaluations. This leads to high overall confidence in approval of biosimilars that successfully complete this assessment. Patients and healthcare providers can expect the same clinical performance of an FDA-approved biosimilar whilst offering the potential to save costs and increase patient access.
Declaration of interest
J Holzmann and J Windisch are employees of Sandoz and S Balser is an employee of Hexal AG. The work was sponsored by Sandoz. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- Kozlowski S, Woodcock J, Midthun K, et al. Developing the nation’s biosimilars program. N Engl J Med. 2011;365(5):385–388.
• First description of the sience of biosimilar development within the regulatory framework required by the BPCI Act.
- Food and Drug Administration. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Silver Spring (MD): U.S. Food and Drug Administration; 2014 May 13.
•• Regulatory guidline which describes the clinical studies needed to support the demonstration of biosimilarity.
- Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. Silver Spring (MD): U.S. Food and Drug Administration; 2015 Apr 28.
•• Regulatory guidline which gives an overview of the FDA’s scientific approach to determine biosimilarity. Describes the totality of evidence concept.
- Food and Drug Administration. Quality considerations in demonstrating biosimilarity to a reference protein product. Silver Spring (MD): U.S. Food and Drug Administration; 2015 Apr 28.
•• Regulatory guidline which describes in great detail which quality factors should be taken into account when analytically characterizing the biosimilar product.
- Food and Drug Administration. Zarxio (filgrastim-sndz) drug approval review documents. 2005 [cited 2015 Nov 14]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/125553Orig1s000TOC.cfm.
• Publicly available document which describes the FDA’s biosimilarity assessment of filgrastim-sndz.
- January 7 2015 Oncology Drugs Advisory Committee Meeting. 2015 [cited 2015 Nov 14]. Available from: http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/ucm426351.htm.
• First Advisory Committee Meeting on a biosimilar.
- Schiestl M, Stangler T, Torella C, et al. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–312.
- CHMP assessment report by the Europeam Medicines Agency for Zarzio. 2008 [cited 2015 Nov 14]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000917/WC500046528.pdf
- Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948–1953.
• Publication which describes the confirmatory clinical trial in breast cancer patients.
- Sörgel F, Schwebig A, Holzmann J, et al. Comparability of biosimilar filgrastim with originator filgrastim: protein characterization, pharmacodynamics, and pharmacokinetics. BioDrugs. 2015;29(2):123–131.
- European Medicines Agency. Questions and answers on biosimilar medicines (similar biological medicinal products). London: European Medicines Agency; 2012.
- Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 2013;72(3):315–318.
• Discusses the subject of manufacturing process changes after approval for mAbs and fusion proteins authorised in rheumatological indications.
- Aapro M, Cornes P, Abraham I. Comparative cost-efficiency across the European G5 countries of various regimens of filgrastim, biosimilar filgrastim, and pegfilgrastim to reduce the incidence of chemotherapy-induced febrile neutropenia. J Oncol Pharm Pract. 2012;18(2):171–179.