
KEYWORDS:
1. An overview of antibody-drug conjugates for non-oncological indications
By definition, antibody–drug conjugates (ADCs) use monoclonal antibodies to selectively deliver potent cytotoxic agent to antigen-expressing tumor cells with reduced off-target toxicity, which has shown clinical success for cancer treatment. Very few applications have been explored outside the field of oncology utilizing non-cytotoxic drugs. In this editorial, we focus on the recent development of ADCs for non-oncological indications such as immune modulation and anti-infection.
Over a century ago, the German scientist Paul Ehrlich proposed the concept of ‘magic bullet’ for tailored and targeted drug delivery in fighting against human diseases, which was considered the prototype for ADCs. In his theory, a toxin could target a disease-causing organism along with an agent selective for that organism.[Citation1] Until recently, two ADCs, Adcetris (brentuximab vedotin for the treatment of Hodgkin’s lymphoma and anaplastic large-cell lymphoma) and Kadcyla (ado-trastuzumab emtansine for the treatment of Her2+ metastatic breast cancer) were approved by Food and Drug Administration in the United States.[Citation2] Although ADCs have shown promises in cancer therapy, very few indications outside oncology have been reported for ADCs.
As an ideal delivery platform, ADCs utilize antibodies as carriers to deliver small molecules selectively to the target, typically followed by efficient internalization and subsequent intracellular drug release. It is worth noting that ADCs are usually described to target antigen-positive cells. However, in reality, ADCs only create a selective exposure to the target cells upon injection. Overall, the selective exposure of small molecules to the desired targets results in significantly improved therapeutic index of the payload. An ADC consists of three building blocks: an antibody carrier, a small molecule payload, and a linker. Each component is critical to the function and efficacy of the ADCs.
For oncological indications, ADCs carry highly cytotoxic agents to selectively kill the target cells. The cytotoxic payloads typically display picomolar to sub-nanomolar IC50 values in a free drug form, e.g. auristatin, maytansine, calicheamicin, duocarmycins, pyrrololoenzodiazepines and α-amanitin.[Citation3] In contrast, for non-oncological indications, the payloads are delivered to the target cells and modulate biological functions without affecting cell viability. These payloads are non-toxin-based and are largely less potent compared to the toxins. Therefore, ADCs for non-oncological applications have a higher standard in terms of selection of targets, antibody carriers, linkers, as well as conjugation approaches to achieve sufficient binding, efficient internalization, and payload release for efficacy. Next we will elaborate these important features using a few successful examples.
In 2012, Moestrup and co-workers first described a biodegradable anti-CD163 ADC that specifically delivers synthetic glucocorticoid to macrophages.[Citation4] The strategy was proposed to deactivate macrophages using glucocorticoids (1, ) by targeting CD163 that is overexpressed on macrophage cells to spare its side effects of systemic exposure. The dexamethasone ADC retains a high binding affinity to CD163 after conjugation, and showed a strong anti-inflammatory effect by reducing tumor-necrosis factor-α (TNF-α) secretion in rat macrophages post-lipopolysaccharide induction. This in vitro activity also translates to its in vivo potency in a rat model. At the equipotent dose, ADCs were demonstrated to mitigate strong systemic adverse effect of dexamethasone including thymus lymphocytes apoptosis, weight loss, and suppression of endogenous cortisol levels.
Figure 1. Structures of antibody-drug conjugates.
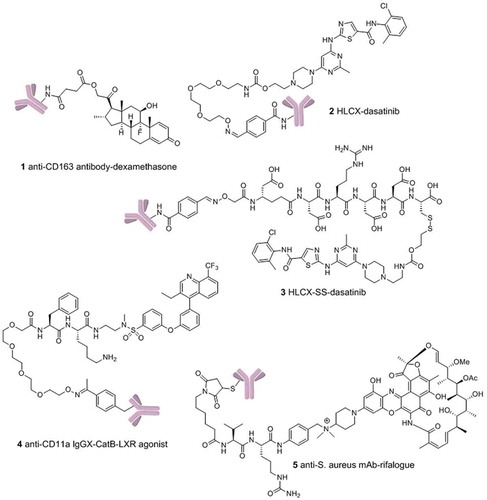
Non-hormone-based immunosuppressants were also used in the treatment of autoimmune diseases such as the kinase inhibitors in the signaling pathways of immune cells. Dasatinib is an oral Bcr-Ab1 tyrosine kinase inhibitor and Src family kinase inhibitor, which is originally developed by Bristol-Myers Squibb for various cancer treatments.[Citation5] Commonly reported side effects include neutropenia, myelosuppression, diarrhea, peripheral edema, and headache.[Citation6] To utilize dasatinib for immune suppression, our group recently reported an immunosuppressive ADC that delivers dasatinib to human T lymphocytes by targeting CXCR4 for the treatment of autoimmune and inflammatory diseases.[Citation7] CXCR4 is highly expressed on T cells, B-cells, and monocytes, as well as hematopoietic stem cells, and has minimal to no expression on non-hematopoietic cells. The anti-CXCR4 antibody (HLCX) used in this work was generated by fusion of a CXCR4 inhibitory peptide into the CDR3H loop of Herceptin using the antibody CDR fusion approach developed in this lab.[Citation8] HLCX is able to engage the ligand-binding pocket through an engineered long hairpin CDR3H loop. Compared to the commercial anti-CXCR4 clone (12G5) that binds to the loop outside the ligand-binding pocket, HLCX showed a much more efficient internalization, which is likely the reason for the potent activity of the resulting ADCs. The linker that tethers the antibody to the small molecule is also critical to the efficacy of ADCs. Sufficient stability is required for ADCs to circulate in the bloodstream without premature drug release. Once internalized, the linker of the ADC should be labile enough to efficiently release the small molecule inside the target cells. In the study of dasatinib ADCs, the antibody was nonspecifically conjugated through lysyl amino groups to dasatinib derivatives via either disulfide-cleavable or noncleavable linkers (2 and 3, ). The resulting ADCs have drug-to-antibody ratios (DAR) around 3. Disulfide bonds are relatively stable in serum and undergo reduction by intracellular glutathione to release the small molecule, while noncleavable linkers rely on intracellular degradation of the antibody. In T-cell activation assays, HLCX-SS-dasatinib was shown to be two-fold more potent than the noncleavable HLCX-dasatinib in suppressing cytokines such as IL-2, TNF-α, and IFN-γ.
While random conjugation generates heterogeneous mixtures of ADCs with varying DARs, site-specific conjugation on the other hand has been shown to improve pharmacokinetics, stability, and drug safety profiles of the ADCs.[Citation9] Scientists at the California Institute for Biomedical Research (Calibr) recently reported an orthogonal amber suppressor tRNA/aminoacyl-tRNA synthetase pair to allow incorporation of p-acetylphenylalanine (pAcPhe) site-specifically into antibodies.[Citation10] This unnatural amino acid-based site-specific conjugation has been applied to an ADC that delivers an LXR nuclear receptor agonist selectively to monocytes/macrophages for the treatment of atherosclerosis (4, ).[Citation11] Liver X receptors are found in macrophages/monocytes as well as kidney, liver, and intestine. To achieve sufficient selectivity, ADC strategy represents a unique approach to selectively deliver LXR nuclear receptor agonist to monocytes/macrophages while minimizing lipogenic effects in hepatocytes. A bio-orthogonal moiety pAcPhe was site-specifically incorporated into Alanine 121 of anti-CD11a IgG. The mutant antibody was then reacted with the terminal aminooxy group on lysosomal protease/cathepsin B (catB) sensitive linker resulting in a DAR of 2. This conjugate (IgGX-CatB-LXR) selectively activated LXR in THP-1 monocyte/macrophage cells (versus HepG2 hepatocytes) with 3-fold greater potency than a known small molecule LXR agonist.
Besides human targets, scientists from Genetech reported an antibody-antibiotic conjugate (AAC) targeting intracellular intracellular S. aureus.[Citation12] The AAC consists of an anti-S. aureus antibody covalently conjugated to a highly efficacious antibiotic via an intracellular protease-sensitive peptide linker, which is cleaved only in the proteolytic environment of the phagolysosome (5, ). This AAC was demonstrated to be superior to vancomycin for the treatment of bacteremia in vivo. To achieve such a high activity, the antibody and antibiotic components were carefully selected to maximize the antibacterial efficacy by targeting intracellular bacteria. Wall-teichoic acids (WTA) are pathogen-specific polyanionic glycopolymers connected to the thick peptidoglycan layers of bacteria. A cathepsin-cleavable linker was applied to tether rifalogue to the anti-WTA antibody covalently for maximal efficacy. A rifamycin derivative (rifalogue) was selected due to its high potency, unaltered bactericidal activity at low pH, and its ability to accumulate intracellularly. On the other hand, rifampicin conjugated to the same anti-WTA antibody showed no potency against intracellular bacteria and exhibited poor in vivo efficacy, which demonstrates the importance of optimizing the payload.
2. Expert opinion
Beyond the cancer therapy, ADCs are getting to show great potential for non-oncological indications. Over the decades, a great number of drug candidates failed in preclinical or clinical stages due to lack of selectivity for desired targets which often results in poor therapeutic index. The ADC strategy thus provides a valuable opportunity for these molecules to be re-evaluated. For example, many potent antibiotic candidates, though capable of killing intracellular pathogens, failed in clinical trials owing to undesired pharmacokinetic profiles or host toxicity. The ADC strategy might overcome these problems through the delivery of potent antibacterial compounds that may not have a suitable profile as unconjugated drugs. Moreover, small molecules that previously failed constitute a great source for ADC development especially for non-oncological indications. In the meantime, for validated targets it may be worth generating new molecules that are specifically designed for non-oncological applications. If the antibody carrier can afford sufficient target selectivity, the selection criteria for payload will mostly be potency. Therefore, re-visiting the hits from earlier screens or re-screening the compound libraries for targets such as kinases and HDAC proteins could provide more and better payloads for non-oncological ADCs. Other than immunological modulation and anti-infection, applications of ADCs for other non-oncological indications such as cardiovascular diseases and liver metabolic disorders are currently undergoing or may be further explored.
Additionally, advancement of ADCs in oncology may shed light on the designing and development of ADCs for non-oncological indications. Perez et al. presented a nice review on the current status and future directions of ADCs.[Citation13] Despite initial success of ADCs for oncology, it remains a great challenge not only to design ADCs, but also to evaluate the efficacy and toxicity of ADCs. Mylotarg (Gemtuzumab ozogamicin), the first ADC to receive marketing approval, was withdrawn owing to a lack of improvement in overall survival in 2010. ADCs for non-oncological applications could face the same challenges. Current disease models may not be sufficient to predict ADC efficacy and toxicity in humans. In addition, it often requires chronic dosing of therapeutics to manage the non-oncological diseases. Therefore, the long-term safety of non-oncological ADCs need to be carefully monitored. Better rodent models are needed to further gauge the effectiveness and risks of the lead ADC molecules to bridge the transition from preclinical studies to clinical trials.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents, received or pending, or royalties.
References
- Ehrlich P. Zur Kenntnis der Antitoxinwirkung. Fortschr Med. 1897;15:41–43.
- Kamath AV, Iyer S. Preclinical pharmacokinetic considerations for the development of antibody drug conjugates. Pharm Res. 2015;32:3470–3479.
- Sassoon I, Blanc V. Antibody-drug conjugate (ADC) clinical pipeline: a review. Methods Mol Biol. 2013;1045:1–27.
- Graversen JH, Svendsen P, Dagnaes-Hansen F, et al. Targeting the hemoglobin scavenger receptor CD163 in macrophages highly increases the anti-inflammatory potency of dexamethasone. Mol Ther. 2012;20:1550−8.
- Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541.
- Giles FJ, O’Dwyer M, Swords R. Class effects of tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia. Leukemia. 2009;23:1698–1707.
- Wang RE, Liu T, Wang Y, et al. An immunosuppressive antibody-drug conjugate. J Am Chem Soc. 2015;137:3229−32.
- Liu T, Liu Y, Wang Y, et al. Rational design of CXCR4 specific antibodies with elongated CDRs. J Am Chem Soc. 2014;136:10557–10560.
- Junutula JR, Raab H, Clark S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925−32.
- Axup JY, Bajjuri KM, Ritland M, et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci U S A. 2012;109:16101–16106.
- Lim RKV, Yu S, Cheng B, et al. Targeted delivery of LXR agonist using a site-specific antibody−drug conjugate. Bioconjugate Chem. 2015;26:2216–2222.
- Lehar SM, Pillow T, Xu M, et al. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature. 2015;527:323–328.
- Perez HL, Cardarelli PM, Deshpande S, et al. Antibody-drug conjugates: current status and future directions. Drug Discov Today. 2014;19:869–881.