Abstract
Neurodegenerative disease such as Alzheimer's, Parkinson and Huntington's are all characterized by dysfunctional neurons and loss of cognitive/motor functions. Interestingly these three diseases involve overproduction, aggregation or abnormal degradation of a specific aberrant protein, which participates in disease pathogenesis. The aggregated proteins may induce disease causing pathways such as high oxidative stress and reduced neuronal metabolism. Several mechanisms are being considered as disease-causing and there is established and growing evidence that a breakdown in neuronal energy production may be an underlying cause in these diseases. The specific risk factors and molecular drivers for each disease vary, yet there are common defective bioenergetics pathways, which may drive neuronal dysfunction. While it has been appreciated that energy deficits can drive neuronal dysfunction and disease, it has for the most part been overlooked as a target pathway for designing novel disease modifying therapeutics. This editorial reviews selected evidence supporting energy deficits as disease-causing and proposes targets for design of new therapeutics.
1. Unique energy demands in neurons
The energy requirement of the brain is disproportionately large compared with other organs, that is, the brain only weighs about 2% of total body weight yet it accounts for 20% of total body energy consumption. These enormous needs are because of specialized functions of neurons such as neurotransmission, neurotransmitter synthesis, maintaining ionic gradients and membrane integrity. The neurons are predominantly dependent on glucose as a substrate for glycolysis and the tricarboxylic acid cycle (TCA) to generate energy (ATP). Any dysfunction in glucose transport, insulin signaling, reduced glycolysis, defective mitochondrial function, such as decreased Krebs cycle activity, and high oxidative stress derails neuronal bioenergetics and results in their dysfunction followed by death. Given that the brain's function and survival are heavily dependent on energy production, any disruption in ATP generation can rapidly turn into disease manifestation.
2. Bioenergetics defects in Alzheimer's disease
Alzheimer's disease (AD) is a chronic and progressive neurodegenerative disorder, which severely effects cognitive function and other behavioral aspects such as executive function and language skills. The disease is characterized by a progressive loss of neurons and synapses.
According to the amyloid hypothesis of AD, accumulation of beta-amyloid (Aβ) peptides as amyloid plaques in the patient's brain is the primary triggering event in the pathogenesis this disease Citation[1]. Both extracellular and intracellular accumulation of Aβ peptides initiates a cascade of events including high oxidative stress, mitochondrial dysfunction and low energy production. A combination of risk factors for the disease such as age, metabolic diseases, genetics and Aβ peptides can ultimately inflict a state of hypometabolism in the brain, that is, reduced ability of this organ to use glucose. Specifically clinical and preclinical data show that AD pathology is accompanied by decrease in activity of enzymes involved in the mitochondrial TCA cycle and oxidative phosphorylation processes, which generate most of the cellular ATP Citation[2]. Some of the molecular changes that can contribute to hypometabolism are decrease in activity of the α-ketoglutarate dehydrogenase complex, a rate-limiting enzyme in the TCA cycle, decreased expression and activity of cytochrome c oxidase,an important component of oxidative phosphorylation via the electron transport chain, and decrease in activity of pyruvate dehydrogenase (PDH), a key rate-limiting enzyme in mitochondria to convert pyruvate into acetyl-CoA, to initiate the TCA cycle Citation[2]. From a viewpoint of bioenergetics based drug discovery, activation of PDH will increase energy production. To achieve this, inhibition of upstream kinases such pyruvate dehydrogenase kinase or glycogen synthase kinase 3β that inactivate PDH could be of therapeutic value in AD.
Human studies also support a defect in energy production and utilization in AD. Evidence suggests that reductions in brain glucose utilization occur at the pre-symptomatic stages of AD Citation[3]. Reduced glucose utilization by the brain has been demonstrated in humans using positron emission tomography (PET) Citation[4]. Such studies have demonstrated consistent, early and progressive reductions in glucose utilization in Alzheimer's patients Citation[5]. Furthermore the extent and regional location of hypometabolism in the brain correlate with severity of symptoms. Overall there is robust preclinical and clinical evidence that poor energy generation may be directly linked to disease genesis in AD Citation[6,7].
3. Bioenergetics defects in Parkinson's disease
Parkinson's disease (PD) is an incapacitating motor-related disease caused by environmental and genetic factors. Clinical symptoms are caused by a loss of dopamine-producing neurons within the substantia nigra region of the brain.
The symptoms of PD are uncontrolled motor deficits such as absence of movement or temporary paralysis also known as akinesia, abnormal slowness of movement or bradykinesia, as well as abnormalities in gait, resting tremor and rigidity. Like in AD where amyloid plaques are visible, there is presence of insoluble cytoplasmic deposits, termed Lewy bodies, in nigral neurons. These bodies are composed of aggregates of α-synuclein, a protein which is the primary culprit in disease genesis and detrimental to neuronal function Citation[8]. The aggregate formation is due to failure of its normal degradation by the lysosomal/proteasome pathway. A combination of environmental factors such as toxic chemicals and α-synuclein can induce mitochondrial dysfunction and oxidative damage as key molecular mechanisms compromising neuronal function. Importantly, they affect mitochondrial electron transport carriers and glucose utilization, which in turn causes energy deficits. Elegant studies using samples from PD patients and systems-biology-based approaches have highlighted specific defects in mitochondrial, electron transport system gene and protein components. It is of significant interest that genes regulated by peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) are downregulated in samples from PD patients Citation[9,10]. PGC-1α is a transcriptional co -activator that interacts with the nuclear receptor PPAR-γ and regulates genes involved in energy metabolism and mitochondrial biogenesis. As such a decrease in its transcriptional activity translates to decreased mitochondrial biogenesis/energy production. PPAR-γ agonists can potentially correct this downregulation of PGC-1α and would be a viable therapeutic approach for PD.
There is also human evidence sustaining a relationship between poor glucose utilization and PD Citation[11]. Magnetic resonance spectroscopy and 2-[18F] fluoro-2-deoxy-d-glucose PET studies demonstrate increased glucose hypometabolism, consistent with a general shift to anaerobic glycolysis in the neocortex of PD patients Citation[12]. Therefore both preclinical and clinical data demonstrate hypometabolism as consistent feature during pathogenesis of PD.
4. Bioenergetics defects in Huntington's disease
Huntington's disease (HD) is an autosomal dominant disease exclusively caused by the expansion of a CAG repeat in the huntingtin (HTT) gene, which encodes a stretch of polyglutamines at the amino terminus. This gene mutation results in production of mutant huntingtin protein (mHtt) which is the primary disease-causing event Citation[13]. Clinically, HD is characterized by motor and cognitive decline including deficits in movement control (chorea, dyskinesias), impairments in executive function, working memory and attention.
HD is characterized by the progressive degeneration of a subset of neurons in the corpus striatum, populations of cortical pyramidal neurons in the motor, frontal, and occipital cortices, as well as neurons in other brain regions such as the hypothalamus. It is becoming increasingly clear that mHtt can decrease mitochondrial function by affecting intracellular calcium levels and mitochondrial membrane potential as well as reducing transcription of key genes Citation[14]. Additionally, the PPAR-γ signaling pathway is impaired by mHtt and activation of this pathway improves mitochondrial deficits. From a therapy perspective, it is of great interest that PPAR-γ agonist rosiglitazone has been shown to increase mitochondrial mass in the striatal cell model of HD and prevent the decrease in mitochondrial membrane potential and the increase in oxidative stress induced by mHtt. mHtt has also been shown to directly decrease the expression of PGC-1α in the striatum of HD mice and samples from HD patients Citation[15]. Based on these observations, mHtt's inhibition of PGC-1α expression and function could be targeted for new therapeutics.
Defective brain energy metabolism has been well documented in HD patients Citation[16]. In patients, there is strong evidence for reduced glucose consumption in the brain, especially in the basal ganglia, even in pre-symptomatic mutation carriers Citation[17]. Thus both preclinical and clinical data support decreased energy production as a putative neuron-impairing event in HD.
5. Conclusions
The discussion above summarizes the potential role of poor energy generation in neurons in AD, PD and HD as disease causing. illustrates how energy failure can occur in each disease. In AD, Aβ peptides together with other risk factors can induce energy failure through some of the events mentioned here. Similarly in PD environmental factors and genetics can work via α-synuclein to cause energy deficits. HD is different in that it's a genetic disease but the detrimental mutant protein mHtt can cause similar mitochondrial and energy failures. Based on the data summarized here as well as other published data we can recommend specific targets for energy improvements. As shown in , for AD, the activation of PDH, a rate-limiting enzyme for Kreb's cycle, can be proposed as a target for drug discovery. For both PD and HD it is possible that targeting PGC-1α may be a viable option. An approach to doing this could be PPAR-γ agonism, which should induce PGC-1α co-activator functions.
Figure 1. The primary aberrant protein drivers and molecular pathways that may be involved in creating energy deficits in neurons triggering dysfunction and disease in AD, PD and HD.
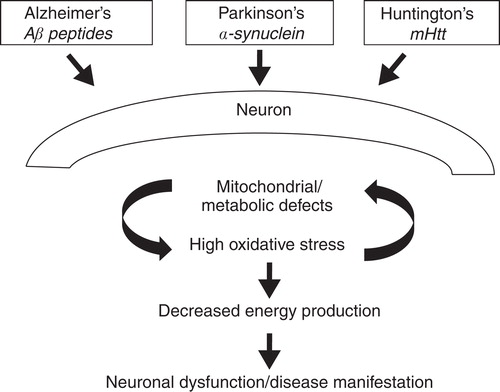
Table 1. Therapeutic targets.
6. Expert opinion
The therapeutics currently available for above mentioned neurodegenerative diseases are purely symptomatic in nature such as the acetylcholine esterase inhibitors for AD and dopamine supplements for PD. While these approaches alleviate the symptoms, they are not disease modifying. The danger of purely relying on such treatments is that while patients may feel better temporarily, the underlying disease continues to progress and get worse. Therefore, new approaches are essential to designing drugs that can block progression of disease and ultimately reverse it.
To this end there has been good progress in the last decade. In AD the focus has been to modulate the amyloid cascade through enzyme inhibitors, vaccines and anti-aggregants. In the case of PD, we are now starting to see approaches directed towardsα-synuclein such as anti-aggregants and for HD very early approaches are aimed at mHtt neuroprotectants and gene therapy.
It is this author's conviction that concomitant with the above disease-modifying approaches we need newer approaches. AD and PD are multifactorial diseases in nature and limiting drug discovery approach to one target is probably too risky and this is reflected in the early data from clinical trails in AD. Addressing a molecular pathway such as neuronal energy production may have a higher likelihood of success because of its direct link with neuronal dysfunction and death. Drugs designed using such an approach can also be used in combination with the existing symptomatic drugs.
Declaration of interest
The author is a compensated advisor to Kareus Therapeutics, SA Co., which is engaged in developing drugs for Alzheimer's and other neurodegenerative diseases.
Bibliography
- Saxena U. Alzheimer's disease amyloid hypothesis at cross roads: where do we go from here? Expert Opin Ther Targets 2010;14:1273-7
- Saxena U. Bioenergetics breakdown in Alzheimer's disease: targets for new therapies. Int J Physiol Pathophysiol Pharmacol 2011;3:133-9
- Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann NY Acad Sci 2008;1147:180-95
- Mosconi L, Mistur R, Switalski R, FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur J Nucl Med Mol Imaging 2009;36:811-22
- Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer's disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging 2005;32:486-510
- Devanand DP, Mikhno A, Pelton GH, Pittsburgh compound B (11C-PIB) and fluorodeoxyglucose (18F-FDG) PET in patients with Alzheimer disease, mild cognitive impairment, and healthy controls. J Geriatr Psychiatry Neurol 2010;23:185-98
- Rostomian AH, Madison C, Rabinovici GD, Jagust WJ. Early 11C-PIB frames and 18-FDG PET measures are comparable: A study validated in a cohort of AD and FTLD patients J Nucl Med. 2011;52:173-9
- Cookson MR, Bandmann O. Parkinson's disease: insights from pathways. Hum Mol Genet 2010;19(R1):R21-7
- Zheng B, Liao Z, Locascio JJ, Global PD Gene Expression (GPEX) consortium. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med 2010;2:52-73
- Ebrahim AS, Ko LW, Yen SH. Reduced expression of peroxisome-proliferator activated receptor gamma coactivator-1alpha enhances alpha-synuclein oligomerization and down regulates AKT/GSK3beta signaling pathway in human neuronal cells that inducibly express alpha-synuclein. Neurosci Lett 2010;473:120-5
- Pappata S, Santangelo G, Aarsland D, Mild cognitive impairment in drug-naive patients with PD is associated with cerebral hypometabolism. Neurology 2011;77:1357-62
- Bohnen NI, Koeppe RA, Minoshima S, Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J Nucl Med 2011;52:848-55
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev 2010;90:905-81
- Damiano M, Galvan L, Deglon N, Brouillet E. Mitochondria in Huntington's disease. Biochim Biophys Acta 2010;1802:52-61
- Kuwert T, Lange HW, Boecker H, Striatal glucose consumption in chorea-free subjects at risk of Huntington's disease. J Neurol 1993;241:31-6
- Grafton ST, Mazziotta JC, Pahl JJ, Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington's disease. Arch Neurol 1992;49:1161-7
- Jenkins BG, Koroshetz WJ, Beal MF, Rosen BR. Evidence for impairment of energy metabolism in vivo in Huntington's disease using localized 1H NMR spectroscopy. Neurology 1993;43:2689-95