
1. Introduction
In the castration-resistant state, androgen receptor (AR)-signaling remains a critical driver of prostate cancer cell growth. This realization led to the development of the next-generation AR-directed agents abiraterone and enzalutamide. It is important to recognize that both of these agents target the ligand-binding domain (LBD) of the AR (abiraterone through ligand depletion and enzalutamide through receptor antagonism), and that only the LBD and DNA-binding domain (DBD) of the AR have been crystalized.[Citation1] Not surprisingly, resistance to abiraterone and enzalutamide is typically heralded by expression of AR-regulated genes such as PSA and the presence of AR protein in the nucleus – indicating persistent activation of the AR transcriptional program. Thus, in spite of best efforts to block AR signaling, the AR is able to bypass these therapies, driving disease progression.
2. AR-splice variants and drug resistance
Several mechanisms of resistance have been identified to current AR-directed therapies, with the majority resulting in re-activation of the AR transcriptional program.[Citation2] Recent RNA sequencing data from large data sets, such as the SU2C International Dream team, strongly suggests that constitutively active AR splice variants (AR-Vs) may play a role in 40–50% of patients castration-resistant prostate cancer (CRPC).[Citation3] These truncated variants of the full-length AR (AR-FL) lack the C-terminal LBD and retain transcriptional activity.[Citation4] Importantly, a subset of the C-terminally deleted variants possesses constitutive activity and appears to be generated in response to various androgen ablation therapies – an effect that is most apparent when the C-terminus is inhibited by potent AR-signaling inhibitors.[Citation2] However, the function of most AR-Vs described has yet to be determined.
Several mechanisms have been described by which variants can be produced. The first, described by Dehm et al., involves rearrangements of the AR gene itself, with AR-V7 (the most prevalent AR-V) and ARv567es generated in response to genomic rearrangements in some cases.[Citation5] The second, being through alternative splicing of mRNA.[Citation6] It seems likely that AR-V7 is primarily produced through a splicing mechanism induced by castration, as this AR-V can be rapidly induced following the inhibition of the AR-signaling program and repressed following AR-FL reactivation through supplementation with androgens.[Citation7]
Preclinical data suggest that targeting the AR-Vs may be an effective strategy given that they function independently of ligand and associate with models of CRPC.[Citation2] Further, the transcriptome generated by AR-Vs is similar to that of liganded AR-FL when they are present as the only receptor; however, they are also associated with a more aggressive cell cycle enhancing program.[Citation8,Citation9] Importantly, AR-V expression increases in CRPC and the presence of their mRNA is associated with decreased PSA response rates and shorter survival on abiraterone and enzlautamide.[Citation3,Citation10] Of note, one key issue that needs to be addressed is the observation that AR-V mRNA expression levels often occur at only ~1% of AR-FL levels, suggesting that they are a minor player in CRPC drug-resistance.[Citation3,Citation10] However, data demonstrating that AR-V7 and ARv567es protein are present in the nucleus bypasses the mRNA argument given that AR is a transcription factor, and chromatin-bound nuclear AR-V protein indicates that it is likely a functional transcription factor.[Citation11,Citation12]
3. Why target AR-Vs?
In one mouse model of CRPC where the AR-Vs are increased, treatment with enzalutamide appeared sufficient to inhibit tumor progression, suggesting that AR-Vs must heterodimerize with AR-FL in order to be activated.[Citation13] However, other similar models demonstrated that neither enzalutamide nor abiraterone could overcome the activity of the constitutively active AR-Vs.[Citation7,Citation9,Citation14–Citation18] Whether this effect is due to the fact that they can homodimerize or heterodimerize with other variants, as has recently been demonstrated, is a possibility.[Citation19]
In 22RV1 cells that express both AR-FL and AR-V7, inhibition of AR-V7 with siRNA to the cryptic exon, CE3, on AR-V7 is necessary to suppress cell proliferation.[Citation20] Thus, supporting the role of AR-Vs as drivers of castration-resistance and indicating that both variant and AR-FLs must be inhibited in order to inhibit tumor growth.
How to target the constitutively active AR-Vs does pose a dilemma (). Although AR-V7 is the most common variant, multiple variants may be responsible for resistance.[Citation3] In which case targeting the unique cryptic exon of AR-V7 may be insufficient to prevent AR-V mediated tumor growth. Certainly, ARv567es (AR-V12) is expressed in a number of enzalutamide resistant tumors. Therefore, either the N-terminal domain or the DBD will need to be targeted in order to effectively inhibit all AR activity. Targeting the N-terminal domain may prove difficult because of its variable structure, however. The N-terminal domains of steroid hormone receptors are intrinsically disordered, and although the folded conformation may be relatively stable – depending on the microenvironment as modified by osmolytes, phosphorylation status, and co-regulatory binding proteins – developing agents that target one-folded conformation may be less effective if binding the N-terminus alters its folding pattern. The possibility therefore remains that even if one form of the N-terminal domain is effectively targeted, others may remain unaffected, thus decreasing the clinical efficacy of an N-terminal domain targeted agent.
Figure 1. Schematic diagram of potential inhibitors of AR-Vs. AR-FL, exon skipping variant ARv567es, and truncation variant AR-V7 proteins are shown. Note each has the transactivation N-terminus encoded by exon 1 (NTD) and the DNA binding domain encoded by exons 2 and 3 (DBD). Only AR-FL and ARv567es contain the hinge region encoded by exon 4 that contains the consensus nuclear localization sequence and poly ubiquination sites. All AR-Vs have a repertoire of co-factors although these have not been fully described for the AR-Vs. Also note that AR-V7 has the cryptic exon, CE3 from intron 3 as its v-terminal exon. As noted in the text, multiple variants have been described and the two AR-Vs shown are representative of the two classes of variants. Potential inhibition points of variant development or activity are indicated and a representative compound(s) that had been reported to act at these sites in included. The compounds listed are not meant to be a comprehensive list at each site since multiple therapeutics for the variants are under development.
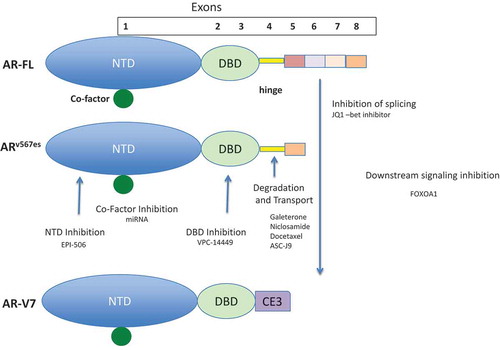
EPI-001 is the first AR N-terminal domain inhibitor. It specifically targets the Tau-5 domain of the AR AF-1 domain within the N-terminus.[Citation21] EPI-001 is a mixture of four stereoisomers, with each stereoisomer binding AR AF-1 at a similar IC-50. EPI-001, its stereoisomers and analogues all effectively inhibit the growth of CRPC xenografts in mice, and NMR studies have confirmed that EPI-001 binds to the Tau-5 domain of the AR. Importantly, it appears effective in preclinical models of AR-V7 positive CRPC and is currently being tested in a Phase I clinical trial in prostate cancer.
Another target that has been suggested is the AR DBD. Rennie et al. have identified a surface exposed pocket on the DBD, which can be targeted by small molecules designed to selectively bind the pocket. These agents are able to effectively block transcriptional activity of both AR-Vs and AR-FL at submicromolar concentrations and inhibit mouse xenografts.[Citation22] This growth inhibition is lost when residues involved in the drug–DBD interaction are mutated – providing evidence that this compound does in fact function through binding the DBD. Further supporting this mechanism of action, the compounds did not affect nuclear localization of the AR, but rather blocked its interactions with chromatin.[Citation22]
Another approach to targeting AR-Vs is to enhance degradation. ASC-J9 was the first compound demonstrated to promote degradation of AR-FL and AR-V7 in preclinical models.[Citation23] Galeterone was originally developed as a CYP-17 inhibitor, and while it does likely inhibit AR-signaling through ligand depletion, it also appears to inhibit AR nuclear translocation and promote AR proteasomal degradation.[Citation24] Preclinical data indicated that this compound may also degrade mutated and AR-V protein through an ubiquination mediated pathway. Of interest, AR-FL receptors do not appear to be degraded following exposure to galeterone. These data suggest that galeterone may be effective against AR-V-induced castration resistance. Galeterone is now being tested in a Phase III trial targeting men with detectable AR-V7 mRNA in circulating tumor cells (NCT02438007).
Niclosamide, an antihelminthic agent, has also been shown to promote AR protein degradation.[Citation25] Also similar to galeterone, niclosamide does not appear to degrade AR-FL – providing a rationale for combining niclosamide with agents that target AR-FL (e.g. abiraterone or enzalutamide). Supporting this hypothesis, preclinical models of CRPC that express AR-V7, and that are otherwise resistant to enzalutamide, demonstrate synergistic growth inhibition when exposed to niclosamide plus enzalutamide.
In addition to targeting AR-Vs themselves, another strategy being pursued involves targeting co-regulators of AR-mediated transcription. Bromodomain 4 is an AR co-activator and inhibiting its function by inhibition of chromatin bing or splicing with small molecules such as JQ1 has proven effective in preclinical models of CPRC.[Citation8,Citation26] A number of additional AR-V co-regulators and downstream AR-V signaling inhibitors (e.g. FOXO1, NF-κΒ, and VAV3) have also been shown in preclinical studies to potentially inhibit AR activity; however, no functional inhibitors have been developed at this point in time.[Citation27–Citation30]
4. Expert opinion
AR-Vs have emerged as one of the most promising biomarkers for resistance to AR-directed therapies. The possibility does remain, however, that the AR-Vs are merely an indicator of a larger resistance program. As outlined, preclinical data strongly supports AR-Vs as drivers of CRPC drug resistance, but these findings do need to be confirmed in clinical samples from patients receiving AR-signaling inhibitors. Thus far, circulating tumor cell derived AR-V mRNA transcripts have been shown to associate with decreased response rates and poor clinical outcomes, and while this finding does support the hypothesis that AR-Vs are drivers of resistance, it is hardly conclusive.
To better determine the biologic relevance of the AR-Vs, drugs that effectively target their activity will have to be tested in randomized clinical trials. In order to more definitively establish AR-Vs as drivers of drug resistance, these studies must show that (1) an AR-V targeted drug is more effective than control (i.e. placebo, active control); (2) AR-V protein is expressed, and the AR-V transcriptional program is active in tumor cells from patients randomized to the control arm; and (3) the AR-V targeted drug effectively inhibits the AR-V transcriptional program. It is through this type of correlative work that the role AR-Vs play in promoting CRPC progression can be determined.
Declaration of interest
This work was supported by NIH PO1-CA090381, DOD PCRP Transformative Award W81XWH-13-2-0093, Veterans Affairs Research Service, and PNW Prostate SPORE-CA097186. SR Plymate has acted as scientific advisor for ESSA Pharma and Astellas Pharma. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- Shaffer PL, Jivan A, Dollins DE, et al. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci U S A. 2004;101:4758–4763.
- Schweizer MT, Yu EY. Persistent androgen receptor addiction in castration-resistant prostate cancer. J Hematol Oncol. 2015;8:128.
- Robinson D, Van Allen EM, Wu Y-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228.
- Kumar R, McEwan IJ. Allosteric modulators of steroid hormone receptors: structural dynamics and gene regulation. Endocr Rev. 2012;33:271–299.
- Li Y, Alsagabi M, Fan D, et al. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011;71:2108–2117.
- Liu LL, Xie N, Sun S, et al. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2014;33:3140–3150.
- Yu Z, Chen S, Sowalsky AG, et al. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res. 2014;20:1590–1600.
- Chan SC, Selth LA, Li Y, et al. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015;43:5880–5897.
- Hu R, Lu C, Mostaghel EA, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–3462.
- Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–1038.
- Liu G, Li A, Sun S, et al. Identification of ARv567es expression profile in the prostate cancer clinical samples with a newly developed antibody [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18–22; Philadelphia (PA): AACR. Cancer Res. 2015;75(15 Suppl). Abstract no. 5159.
- Efstathiou E, Titus M, Wen S, et al. Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur Urol. 2015;67:53–60.
- Watson PA, Chen YF, Balbas MD, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–16765.
- Mostaghel EA, Marck BT, Plymate SR, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–5925.
- Dehm SM, Schmidt LJ, Heemers HV, et al. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477.
- Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313.
- Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22.
- Sun S, Sprenger CCT, Vessella RL, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730.
- Xu D, Zhan Y, Qi Y, et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015;75:3663–3671.
- Li Y, Chan SC, Brand LJ, et al. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–489.
- Myung J-K, Banuelos CA, Fernandez JG, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948–2960.
- Li H, Ban F, Dalal K, et al. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J Med Chem. 2014;57:6458–6467.
- Yamashita S, Lai K-P, Chuang K-L, et al. ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia. 2012;14:74–83.
- Yu Z, Cai C, Gao S, et al. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin Cancer Res. 2014;20:4075–4085.
- Liu C, Lou W, Zhu Y, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. 2014;20:3198–3210.
- Asangani IA, Wilder-Romans K, Dommeti VL, et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol Cancer Res. 2016. [Epub ahead of print].
- Bohrer LR, Liu P, Zhong J, et al. FOXO1 binds to the TAU5 motif and inhibits constitutively active androgen receptor splice variants. Prostate. 2013;73:1017–1027.
- Nadiminty N, Tummala R, Liu C, et al. NF-κB2/p52 induces resistance to enzalutamide in prostate cancer: role of androgen receptor and its variants. Mol Cancer Ther. 2013;12:1629–1637.
- Peacock SO, Fahrenholtz CD, Burnstein KL. Vav3 enhances androgen receptor splice variant activity and is critical for castration-resistant prostate cancer growth and survival. Mol Endocrinol. 2012;26:1967–1979.
- Mediwala SN, Sun H, Szafran AT, et al. The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate. 2013;73:267–277.