
ABSTRACT
Introduction: Chronic pain is a massive clinical problem. We discuss the potential of subtype selective sodium channel blockers that may provide analgesia with limited side effects.
Areas covered: Sodium channel subtypes have been linked to human pain syndromes through genetic studies. Gain of function mutations in Nav1.7, 1.8 and 1.9 can cause pain, whilst loss of function Nav1.7 mutations lead to loss of pain in otherwise normal people. Intriguingly, both human and mouse Nav1.7 null mutants have increased opioid drive, because naloxone, an opioid antagonist, can reverse the analgesia associated with the loss of Nav1.7 expression.
Expert Opinion: We believe there is a great future for sodium channel antagonists, particularly Nav1.7 antagonists in treating most pain syndromes. This review deals with recent attempts to develop specific sodium channel blockers, the mechanisms that underpin the Nav1.7 null pain-free phenotype and new routes to analgesia using, for example, gene therapy or combination therapy with subtype specific sodium channel blockers and opioids. The use of selective Nav1.7 antagonists together with either enkephalinase inhibitors or low dose opioids has the potential for side effect-free analgesia, as well as an important opioid sparing function that may be clinically very significant.
KEYWORDS:
1. Introduction
Human-validated analgesic targets such as the sodium channels Nav1.7, Nav1.8 and Nav1.9 are of great interest for the development of new pain therapies and are the topic of the present review. Pain severely afflicts about half a billion people on the planet but has not seen the remarkable progress in treatment that other areas of medicine such as cardiovascular disease or cancer have undergone. One reason for this is that we know very little about the mechanisms that underlie different sorts of pain. Genetic analyses of mouse loss-of-function mutants, particularly tissue-specific knock-outs, suggest that there are many distinct cellular and molecular mechanisms that can give rise to apparently similar pain conditions, such as mechanical, thermal or cold allodynia, where innocuous stimuli cause pain.[Citation1] In humans, major efforts to phenotype neuropathic pain patients and examine different drug regimens are paying dividends, but we still have a limited knowledge of the types of sensory neurons involved in different human pain conditions, let alone the central mechanisms that modulate pain or the location of pain sensations. Given this ignorance, blocking peripheral nerves as a route to treating many different types of pain is attractive. Nerve block has been used for decades as an effective treatment for most pain conditions and relies upon suppressing the electrical signals carried by voltage-gated sodium channels.[Citation2–Citation4] Molecular cloning techniques have revealed nine related voltage-gated sodium channels with distinct biophysical properties, interacting proteins and cellular patterns of expression that are involved in electrical signaling. If specific sodium channels subtypes are involved in particular pain mechanisms, subtype-specific sodium channel antagonists could, in theory, produce side effect-free pain treatment. This has been the goal of many research groups over the past two decades.
2. Genetically defined sodium channel targets
In the post genomic era it has become straightforward to identify the genes linked to human monogenic disorders, and to produce transgenic models in mice for mechanistic studies. These approaches have been particularly fruitful in the study of the role of sodium channels in pain processing. The three sodium channels Nav1.7, Nav1.8 and Nav1.9 are predominantly associated with peripheral neurons rather than central neurons and have all been linked to human monogenic pain disorders.[Citation5,Citation6] The encoding genes, main anatomical expression sites, involvement in diseases/syndromes and pharmacological and electrophysiological features of these three channels are displayed in .
Table 1. Voltage-gated sodium channel α-subunits: types, encoding genes, main anatomical expression sites, involvement in diseases/syndromes, pharmacological and electrophysiological features.
3. Nav1.7 dependent and independent pain states
The first evidence that Nav1.7 was important in peripheral pain pathways came from a conditional knockout study in a subset of mouse sensory neurons expressing another sodium channel, Nav1.8.[Citation13] These sensory neurons are known to be important for inflammatory pain, and the conditional deletion of Nav1.7 in these cells produced a dramatic loss in inflammatory pain.[Citation13,Citation14] In 2004 a Chinese group identified mutations in Nav1.7 in humans suffering from inherited erythromelalgia (IEM) which is a chronic inflammatory condition characterized by pain attacks.[Citation15] The mechanism underlying this condition was unraveled by the laboratory of Stephen Waxman who showed that a large number of different IEM-associated mutations all lead to increased excitability of Nav1.7.[Citation16,Citation17] Another related gain-of-function human pain condition, originally defined as familial rectal pain (FRP) and subsequently renamed paroxysmal extreme pain disorder (PEPD), maps to mutations in the region of Nav1.7 involved in channel inactivation.[Citation18] This disorder is associated with excruciating mechanically evoked pain. Much effort has been made to try and underpin the mechanistic changes in Nav1.7 channel function that give rise to IEM and PEPD. It has been hypothesized that IEM is principally caused by a shift in channel activation, whereas PEPD is caused by a shift in channel inactivation. This hypothesis was further supported by the discovery of a mutation that causes changes in both activation and inactivation kinetics of Nav1.7, which subsequently results in a clinical phenotype that is indicative of both IEM and PEPD.[Citation19] Furthermore, a link between enhanced resurgent currents in PEPD mutations but not IEM-linked mutations has also been noted.[Citation20] More recently several IEM-causing mutations have been discovered that do not have the characteristic shift in channel activation, suggesting that the etiology of these pain disorders, particularly IEM, is more complex than first thought.[Citation21,Citation22] Such human gain-of-function pain-related mutations in Nav1.7 have stimulated considerable interest in the pharmaceutical industry.
In 2006 James Cox and Geoff Woods found that loss-of-function recessive mutations in Nav1.7 resulted in congenital insensitivity to pain (CIP).[Citation23] This dramatic discovery energized the field to focus on this particular sodium channel isoform for the development of new analgesic drugs that should, in principal, be side-effect free. As global sodium channel blockers are effective analgesics, a critical issue in the development of such drugs is a demonstration of specificity for Nav1.7, a vital element that is lacking in many Nav1.7 drug development programs. Patents filed in the area have recently been reviewed [Citation24], whilst clinical trial data are summarized in .
Table 2. Voltage-gated sodium channel inhibitors in current study.
Importantly, although acute pain and some types of inflammatory and neuropathic pain appear to be Nav1.7 dependent, not all pain states are dependent on Nav1.7. Recently, examples of pain states that are not dependent upon the expression of Nav1.7 have been identified in both mice and humans. In mice, bone cancer pain and oxaliplatin-evoked mechanical and cold allodynia all occur normally in Nav1.7 null mutant mice.[Citation1] In humans a recent case report suggests that individuals who carry loss-of-function mutations in SCN9A, associated with CIP, still have the potential of developing neuropathic pain.[Citation39] Thus Nav1.7-targeted antagonists are not the panacea for all pain syndromes, despite the remarkably broad role of the channel in acute and inflammatory pain states.
Given the fact that many Nav1.7 drug development programs have been underway for several years, success has been limited. Potent specific stable antagonists have been developed and tested in humans (see ). Disappointingly, a recent claim that neutralizing monoclonal antibodies to Nav1.7 are effective analgesics has not been replicated.[Citation40] Why has this promising area of drug development apparently as yet failed to produce good analgesics? The impression gained is that the more selective an inhibitor is for Nav1.7 (e.g. protoxin II), the less potent the analgesia, whilst less selective antagonists (e.g. CNV-1014802 and lidocaine) that may exert effects on a broader spectrum of sodium channels are very effective.
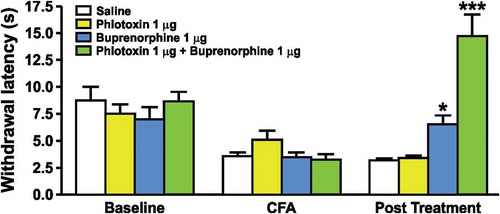
An explanation for this conundrum comes from the surprising discovery that there is a major role for enhanced opioid signaling in the analgesia associated with Nav1.7 null mutant CIP. Studies on CIP patients that were potentially Nav1.7 null mutants in the pre-genomic era had already provided evidence that the endogenous opioid system contributed substantially to the pain free state.[Citation41] When analgesia is established by the deletion of SCN9A encoding Nav1.7 in mice, the vast majority of analgesia is naloxone reversible. In a single human Nav1.7 null subject, noxious stimuli could be detected 80% of the time after naloxone treatment, but not before.[Citation42] In other words, opioid-mediated analgesia seemingly accounts for most of the hypoalgesic phenotype of Nav1.7 null mutant mice and humans. Loss of Nav1.7 expression is linked to a transcriptional upregulation of Penk, the precursor of met-enkephalin, that is found at high levels in the central terminals of Nav1.7 null sensory neurons.[Citation42] Complete channel block in wild type DRG neurons in culture with high levels (0.5 µM) of tetrodotoxin (TTX), a sodium channel pore blocker [Citation42], also leads to upregulated expression of opioid peptides in sensory neurons. However, TTX at five times the IC50 for Nav1.7 does not lead to enhanced enkephalin expression, suggesting that any compound that recapitulates the CIP phenotype of loss-of-function mutants will have to provide 100% Nav1.7 channel block, which is an unrealistic pharmacological goal. As opioid-dependent analgesia seems to account for the vast majority of the CIP phenotype, intriguingly implying a life-long endogenous opioid action with no tolerance [Citation42], a combination of a specific Nav1.7 antagonist and low doses of opioids or enkephalinase blockers should recapitulate CIP if this mechanism is correct. In animal models, this conclusion has been confirmed for a number of acute, inflammatory and neuropathic pain models.[Citation1,Citation43,Citation44] In , the combination of a selective toxin that blocks Nav1.7, phlotoxin 1, with buprenorphine at concentrations that are ineffective alone produces a dramatic analgesia when applied together (Patent number: WO2015036734). The development of new enkephalinase inhibitors [Citation45] provides an alternative strategy of combining enkephalinase inhibitors and Nav1.7 antagonists to cause analgesia.
Figure 1. The effect of phlotoxin and/or buprenorphine on the heat hyperalgesia induced by injection of CFA on the hind paw of mice. The latency of paw withdrawal in response to a nociceptive heat stimulus (Hargreaves test) was evaluated before (baseline) and 24 hours after the intraplantar injection of CFA with (post-treatment) or without (CFA) the administration of phlotoxin and/or buprenorphine (30 minute administration). Each group is represented by a different coloured bar (saline – white; phlotoxin – yellow; buprenorphine – blue; phlotoxin + buprenorphine – green) with the administration of test compounds only being summarized in the post-treatment bars. Values represent means ± SEM of 6–8 mice. *p < 0.05 and ***p < 0.001 when compared to saline group (one-way ANOVA followed by Bonferroni post hoc test).
How does the presence of a voltage-gated sodium channel influence the expression of opioid peptides? This is a fascinating mechanistic puzzle. Importantly, altering intracellular calcium levels does not seem to link sodium channel activity and enkephalin expression.[Citation42] In contrast, manipulating intracellular sodium levels can alter expression of the penk mRNA that produces leu and met-enkephalins; the sodium ionophore monensin down-regulates expression, whilst channel block with very high dose TTX upregulates penk mRNA.[Citation42] Sodium thus seems to be functioning as a second messenger, and this parallels the situation in the kidney where tonicity regulates gene expression through effects on salt kinases and a transcription factor NFAT5, that is also expressed at very high levels in sensory neurons.[Citation46] This potential mechanism is an area of research interest. Should this mechanism be at play, it is hard to understand why it is linked to voltage-gated Nav1.7 channel activity and not to other sodium channels such as Nav1.8 that are present in the same cells. A possible explanation is that sodium ingress through the Nav1.7 window current has a much greater effect on intracellular sodium concentrations than any other sodium channels. Consistent with this hypothesis, HEK293 cell lines permanently expressing Nav1.7 have resting intracellular sodium levels that are double the level of the parental cell line (data not shown). This could explain a specific link between persistent Nav1.7 channel activity and substantial changes in intracellular sodium concentrations that may have effects as a second messenger. Nav1.9 window currents are also substantial, but loss of this channel does not alter penk expression.[Citation42] Thus the link between intracellular sodium levels and penk expression remains uncertain, although channel subcellular localization as well as expression may be an important aspect of such potential signaling mechanisms.
4. Nav1.8
The role of Nav1.8 in nociceptive processing has been extensively studied, with numerous behavioral and functional studies underlining the importance of Nav1.8 channels, as well as Nav1.8-expressing neurons, in the development of inflammatory and neuropathic pain conditions.[Citation14,Citation47–Citation50] These studies have highlighted the potential impact of targeting Nav1.8 for treating numerous pain conditions; however, in contrast to SCN9A, naturally occurring loss-of-function mutations occurring in SCN10A are yet to be described in humans, and therefore the therapeutic potential of targeting Nav1.8 has to be extrapolated from studies conducted on mice. Importantly, however, several gain-of-function mutations have been reported for SCN10A, which strongly support a role of Nav1.8 in nociceptive processing in humans. Recent genetic analysis of 104 patients with idiopathic painful neuropathy, for which mutations in SCN9A had been ruled out, identified seven mutations in SCN10A in nine individuals.[Citation51] From the seven mutations identified, Faber et al. (2012) identified two gain-of-function mutations in SCN10A (L554P and A1304 T) which altered the gating properties of Nav1.8 and led to an increase in excitability in small neurons. Other gain-of-function mutations in SCN10A have been reported and are also associated with painful neuropathy (predominantly small fiber neuropathy) caused by alterations in channel gating that promote neuronal hyperexcitability.[Citation52,Citation53] Currently there are no Nav1.8-specific compounds in clinical testing; however, there are several compounds that have been shown to be efficacious in animals models of inflammatory, and perhaps more surprisingly, neuropathic pain.[Citation54,Citation55]
Besides nociception, Nav1.8 has also been proposed to play a significant role in cardiac electrophysiology, being expressed in intracardiac neurons where it acts to prolong the PR-interval (atrioventricular conduction) of the cardiac action potential.[Citation56] A genome-wide association study (GWAS) published in 2010 showed that genetic variations in SCN10A can ultimately influence cardiac conduction.[Citation54] Chambers et al. (2010) associated a nonsynonymous short nucleotide polymorphism (SNP) in SCN10A with prolonged atrioventricular conduction, predisposing affected individuals to a higher risk of heart block. Similar association studies have also identified a similar link between genetic variants in SCN10A and atrioventricular conduction properties as well as atrial fibrillation, adding further support for a significant role of Nav1.8 in cardiac electrophysiology.[Citation57–Citation59] Although the deletion or inhibition of Nav1.8 does not seem to adversely affect cardiac output in mice, the role of Nav1.8 in cardiac conduction will nevertheless be an important consideration when developing potential analgesics.[Citation54,Citation60]
5. Nav1.9
In animal models of inflammatory pain the participation of Nav1.9 sodium channels has been well established. Many papers show a reduction in the pain behavior by inflammatory agents such as formalin, carrageenan, CFA [Citation61,Citation62], prostaglandin E2 [Citation63], bradykinin, serotonin and ATP [Citation64] in Nav1.9 knockout mice. The correlation of Nav1.9 sodium channel activity with nerve injury-induced pain is still somewhat uncertain in mouse models. Nav1.9-null mice showed unaltered pain-related behavior in various neuropathic pain models, including partial sciatic nerve injury [Citation64], chronic constriction injury [Citation65] and spinal nerve transaction.[Citation1] However, there was a significant reduction in slowly inactivating and persistent TTX-resistant currents in L4/5 DRG after transection of the sciatic nerve.[Citation66] Furthermore, orofacial neuropathic pain produced by constriction of the infraorbital nerve in mice is dependent on the presence of Nav1.9.[Citation67]
The presence of seven different mutations in the SCN11A gene encoding Nav1.9 channels in peripheral neuropathy patients confirm its participation in neuropathic pain in humans. Two of those mutations (I381T and L1158P) led to a reduction in the current threshold and increased firing frequency in response to suprathreshold stimuli, resulting in increased excitability of DRG neurons.[Citation68] Zhang et al. (2013) also described two mutations in the SCN11A gene (R225C and A808G) in patients experiencing episodic chronic pain.[Citation69] Another Nav1.9 mutation, G699R, which is located in the DII/S4-5 linker, has been identified in a patient with symptoms of painful small fiber neuropathy. The G699R mutant channels render DRG neurons hyperexcitable.[Citation70] More recently, a new gain-of-function mutation in the SCN11A gene (p.V1184A) has been linked to enhanced cold pain in humans.[Citation7] Furthermore, an intriguing observation correlates an unusual syndrome of loss-of-pain sensation and inclination for self-mutilation with a mutation in SCN11A (L811P), which is associated with a gain of function in Nav1.9 sodium channel activity.[Citation71] Other Nav1.9 mutations have recently been linked to enhanced cold pain in humans.[Citation7]
6. Multiple functions for sodium channels
Action potential propagation by sodium channels has long been the principal interest of electrophysiologists. However, increasing evidence links sodium channels to a variety of other functions in both neurons and supposedly non-excitable cells. Thus both Nav1.5 and Nav1.7 expression have been linked to the ability of cancer cells to metastasize.[Citation72] In the pain field, the ability of sympathetic neurons to form baskets around sensory neurons in cell bodies and sensitize peripheral pain pathways is dependent on the expression of Nav1.7 in the sympathetic neurons.[Citation1] The mechanisms underlying these events are uncertain. One suggestion has been that accessory beta subunits with their cell adhesion motifs are involved in cell migration.[Citation73] An alternative suggestion has been that sodium channel expression increases baseline intracellular sodium levels, and sodium proton anti-porters acidify the extracellular milieu allowing cells to penetrate surrounding tissue more effectively.[Citation74,Citation75] These suggestions have yet to be formally proved, and other mechanisms may be at play.
More recently, a link has been made between sodium channel activity and transcriptional regulation, and a possible role for sodium as a second messenger has been described in sensory neurons.[Citation42] The sodium channel Nav1.3 plays an important role in the pancreas in terms of regulating insulin secretion, whilst Nav1.7 has a very broad array of functions, including control of neurotransmitter release in olfactory sensory neurons, as well as regulation of peptide secretion in the hypothalamus.[Citation76–Citation79] All of these functions may create some difficulties with respect to the effective use of sodium channel blockers as side effect-free analgesics.
7. Small molecule blockers of sodium channels as analgesics
Although Nav1.7 is currently one of the most promising targets for alleviating chronic pain, progress on the development of new blockers is intrinsically linked to achieving high levels of selectivity and efficacy. Currently, the majority of therapeutically used sodium channel blockers bind to highly conserved residues that are found within the pore domain of the channel, making selectivity between family members difficult to achieve. These functionally selective blockers often rely upon the channel to enter particular states (typically active, inactive or resting) in order for them to reach their binding site within the inner vestibule of the channel pore. One way of improving selectivity is to design compounds that bind to areas outside of the pore-forming region that are poorly conserved between family members. These compounds are often termed molecularly selective as their inhibitory action is independent of the channel’s functional state.[Citation24] One such compound, PF-05089771 (Pfizer), is currently in clinical trials for use in chronic pain. This molecularly selective aryl sulfonamide compound boasts 1000-fold selectivity for Nav1.7 over Nav1.5 and Nav1.8, and has been reported to be well tolerated in phase I trials.[Citation80] Interestingly, from the information that is currently available, sulfonamides (particularly aryl sulfonamides) seem to be one of the principal classes of compounds used in the development of Nav1.7 inhibitors, suggesting that these compounds may offer a selective advantage over other classes.[Citation24] There are, however, other compound classes in clinical development including the pyrrolidine-based compound CNV-1014802 (convergence), which is currently undergoing phase III clinical trials for use in trigeminal neuralgia.[Citation28] Unfortunately there is currently no information on how selective this compound is over other Nav family members, or indeed where the compound binds the channel . A summary of Nav-specific compounds currently undergoing clinical assessment for treating pain is shown in ; however, owing to the lack of disclosed information, it is difficult to assess the relative selectivity of many of these compounds.
In addition to small molecule inhibitors, several natural toxins are also being exploited for their potential therapeutic benefit. Numerous examples are available, with peptide toxins extracted from tarantula venom (protoxin II) or the venom of the cone snail (µ-Conotoxin – KIIIA) showing reasonable levels of specificity against Nav1.7.[Citation38] Another natural toxin that is being investigated for use in treating pain is tetrodotoxin (TTX), the guanidine-related venom extracted from the puffer fish. TTX shows very little selectivity across a number of Nav family members, with IC50 values for Nav1.1, 1.2, 1.3, 1.4, 1.6 and 1.7 being in the single nanomolar range.[Citation38] Despite the lack of selectivity, TTX is currently undergoing phase III clinical trials for treatment in cancer-related pain, where it is administered subcutaneously to limit systemic effects.[Citation81] Although the selectivity and therapeutic index of natural toxins may limit their therapeutic use, they hold promise as scaffolds for the development of more specific inhibitors targeting for example, Nav1.7.
8. Gene therapy focused on sodium channels
Gene therapy has made enormous strides recently, so that it is at last the focus of interest of reputable groups. AAV mediated gene delivery is of particular interest, but the irreversible silencing of sodium channel genes is potentially problematic. Many genes, as we have seen, have a variety of functions in both neuronal and non-neuronal tissues, and AAV is not neuron specific. Ideally reversible gene therapy with a drug inducible promoter driving antisense constructs or siRNAs could obviate many potential problems associated with a complete irreversible knock down of channel expression. How could this be achieved? A number of approaches have been investigated. The Tet-on system has been examined thoroughly using doxyclin in rodents and primates, but the development of an immune response to components of the viral delivery system are still impeding progress. Drug regulated control of gene expression is a vast prize in terms of general utility for many patients if such technical obstacles can be overcome, but as yet, they have not.[Citation82] A more recent approach that has worked in models of epilepsy exploits a designer receptor activated by a designer drug (DREADD) delivered with AAV. Application of the DREADD activator effectively silenced the epileptic activity [Citation83] suggesting that a similar approach could be effective in pain. Interestingly, an antisense transcript is found for Nav1.7, but its physiological role and significance remain to be comprehensively explored.[Citation84]
9. Expert opinion
Three points are worth making. First, the promise of a side-effect free sodium channel blocking analgesic has yet to be fulfilled, despite the clear utility of nerve block in pain treatment. One reason for this is that Nav1.7 is both a conduit for electrical signaling, as well as a regulator of opioid activity in mice and humans.[Citation42] Complete channel block, mimicked in null mutants, appears to be required for upregulated opioid activity, and this is currently not achieved by small molecules at acceptable concentrations. These observations underscore the essential role for mouse mechanistic studies in human drug development. Such information also point the way forward to effective strategies for treating pain using combination therapy that is very effective in animal models, but requires confirmation with human data that should soon be available.
Secondly, a reason for the failure to develop useful analgesics results from semantic confusion over central versus peripherally acting drugs. Peripheral sensory neurons have terminals in the spinal cord within the blood brain barrier (BBB). These terminals have high concentrations of Nav1.7 protein that is involved in neurotransmitter release. Thus BBB permeant Nav1.7 blockers are essential to block all aspects of Nav1.7 function, even though Nav1.7 is a nominally peripheral neuron-associated protein. It is important to remember that even non-steroidal anti-inflammatory drugs (NSAIDS) that are assumed to work peripherally through the blockade of sensitizing cyclooxygenase metabolites, such as prostaglandins, are highly effective when delivered intrathecally, suggesting that actions of neuronal cyclooxygenase metabolites on the central terminals of sensory neurons are of great importance in inflammatory pain. Thus peripheral neuron-targeted drugs may need to be BBB permeant to affect their actions.
Finally, the great advances made in whole genome sequencing, and the claims of some of the functional imaging community has led to the specious claim that drug development work can be carried out without animal studies. This is dangerously naive. Genetic manipulation in mice gives us the mechanistic insights that allow rational drug design, as demonstrated emphatically by the present Nav1.7 antagonist analysis. Of course there are differences between mice and humans, but many more examples of drug failure between phase 2 and 3 can be identified than those that occur as a result of species differences. An investment in basic mechanistic research is the key to new drugs, whilst the best medicinal chemistry focused on a poorly understood target is likely to fail.
Article highlights
Non-specific sodium channel blockers are very effective analgesics for most pain syndromes.
Sodium channel Nav1.7 is essential for human pain, but specific antagonists have weak analgesic activity.
Nav1.7 not only propagates action potentials but has other actions, notably in control of the expression of opioid peptides.
Animal models show that Nav1.7 antagonists, when co-administered with low dose opioids do give effective analgesia.
The role of Nav1.8 and Nav1.9 in pain syndromes is explored.
Small molecule blockers and gene therapy approaches to down-regulating Nav1.7 sodium channel expression are described.
This box summarizes the key points contained in the article.
Declaration of interest
E Emery and J Wood thank the Wellcome Trust, J Wood thanks the Medical Research Council and A Luiz and J Wood thank Arthritis UK. J Wood also thanks the BK21 Programme at SNU for generous support. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Acknowledgement
The authors gratefully acknowledge the generous gift of Phlotoxin by Dr Pierre Escoubas.
References
- Minett MS, Falk S, Santana-Varela S, et al. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. 2014;6(2):301–12.
- Dobney GH, Belam OH. The scope of nerve root block in physical medicine. Proc R Soc Med. 1963;56:444–6.
- Bonica JJ. Management of pain with regional analgesia. Postgrad Med J. 1984;60(710):897–904.
- Fritz J, Chhabra A, Wang KC, et al. Magnetic resonance neurography-guided nerve blocks for the diagnosis and treatment of chronic pelvic pain syndrome. Neuroimaging Clin N Am. 2014;24(1):211–234.
- Eijkelkamp N, Linley JE, Baker MD, et al. Neurological perspectives on voltage-gated sodium channels. Brain. 2012;135(Pt 9):2585–2612.
- Dib-Hajj SD, Black JA, Waxman SG. Nav1.9: a sodium channel linked to human pain. Nat Rev Neurosci. 2015;16(9):511–519.
- Leipold E, Hanson-Kahn A, Frick M, et al. Cold-aggravated pain in humans caused by a hyperactive Nav1.9 channel mutant. Nat Commun. 2015;6:10049.
- Wood JN, Baker M. Voltage-gated sodium channels. Curr Opin Pharmacol. 2001;1(1):17–21.
- Ogata N, Ohishi Y. Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn J Pharmacol. 2002;88(4):365–377.
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edition. Br J Pharmacol. 2009;158:S1–S239.
- Lees G, Shipton E. Voltage-gated sodium channels in nociception and their potential as targets for new drugs in treatment of chronic neuropathic pain. Curr Anaesthesia Crit Care. 2009;20(5–6):204–208.
- Vickery RG, Amagasu SM, Chang R, et al. Comparison of the pharmacological properties of rat Na(V)1.8 with rat Na(V)1.2a and human Na(V)1.5 voltage-gated sodium channel subtypes using a membrane potential sensitive dye and FLIPR. Recept Channels. 2004;10(1):11–23.
- Nassar MA, Stirling LC, Forlani G, et al. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A. 2004;101(34):12706–12711.
- Abrahamsen B, Zhao J, Asante CO, et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321(5889):702–705.
- Yang Y, Wang Y, Li S, et al.. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41(3):171–174.
- Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci. 2004;24(38):8232–8236.
- Waxman SG. Painful Na-channelopathies: an expanding universe. Trends Mol Med. 2013;19(7):406–409.
- Fertleman CR, Baker MD, Parker KA, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774.
- Estacion M, Harty TP, Choi J-S, et al. A sodium channel gene SCN9A polymorphism that increases nociceptor excitability. Ann Neurol. 2009;66(6):862–866.
- Theile JW, Jarecki BW, Piekarz AD, et al. Nav1.7 mutations associated with paroxysmal extreme pain disorder, but not erythromelalgia, enhance Navbeta4 peptide-mediated resurgent sodium currents. J Physiol. 2011;589(Pt 3):597–608.
- Eberhardt M, Nakajima J, KLinger AB, et al. Inherited pain: sodium channel Nav1.7 A1632T mutation causes erythromelalgia due to a shift of fast inactivation. J Biol Chem. 2014;289(4):1971–1980.
- Emery EC, Habib AM, Cox JJ, et al. Novel SCN9A mutations underlying extreme pain phenotypes: unexpected electrophysiological and clinical phenotype correlations. J Neurosci. 2015;35(20):7674–7681.
- Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–8.
- Sun S, Cohen CJ, Dehnhardt CM. Inhibitors of voltage-gated sodium channel Nav1.7: patent applications since 2010. Pharm Pat Anal. 2014;3(5):509–521.
- Convergence Pharmaceuticals, C., UK. Convergence Pharmaceuticals’ Novel Nav1.7 Selective Sodium Channel Blocker CNV1014802 Demonstrates Proof of Concept in Second Neuropathic Pain Study in Lumbosacral Radiculopathy. [ updated 2014 Sep 23; [cited Jan, 2016] Available from: http://www.convergencepharma.com/userfiles/file/140923_LSR_FINAL.pdf.
- Convergence Pharmaceuticals, C., UK. CNV1014802. 2016 [cited 2016 Jan] Available from: http://www.convergencepharma.com/index.asp?page_id=14.
- Convergence Pharmaceuticals, C., UK. Convergence pharmaceuticals receives orphan-drug designation for Nav1.7 blocking pain drug CNV1014802. [ updated Jul 30 2013; [cited 2016 Jan] Available from: http://www.convergencepharma.com/userfiles/file/Orphan%20Drug%20Designation_Final.pdf.
- Convergence Pharmaceuticals, C., UK. The Pipeline. 2016 [cited 2016 Jan] Available from: http://www.convergencepharma.com/?page_id=16.
- Convergence Pharmaceuticals, C., UK. A Phase I ready novel sodium channel blocker CNV1061436 for CNS disorders is added to the pipeline of clinical development candidates. [ updated 2013 Oct 29; cited 2016 Jan] Available from: http://www.convergencepharma.com/userfiles/file/131029%20Convergence_%20436%20press%20release%20final%20complete.pdf.
- Convergence Pharmaceuticals, C., UK. Convergence presentations and posters (preclinical properties of CNV3000223, a new Nav1.7 selective blocker with broad anti-hyperalgesic efficacy). 8th Congress of the European Federation of IASP Chapters (EFIC) - Pain in Europe V; 2013 Oct 9–12; Florence, Italy. 2016; http://www.convergencepharma.com/index.asp?page_id=29 [cited 2016 Jan].
- Xenon Pharmaceuticals Inc., V., Canada. Xenon’s XEN402 ointment significantly relieves pain in patients with post herpetic neuralgia. [ updated 2011 May 2;cited 2016 Jan]. Available from: http://www.xenon-pharma.com/2011/05/xenon%E2%80%99s-xen402-ointment-significantly-relieves-pain-in-patients-with-post-herpetic-neuralgia/.
- Xenon Pharmaceuticals Inc., V., Canada. TV-45070 for the treatment of pain. 2015 [cited 2016 Jan] Available from: http://www.xenon-pharma.com/product-candidates/pain/pain-teva/.
- Genentech. Pipeline. 2016 [cited 2016 Jan] Available from: http://www.gene.com/medical-professionals/pipeline.
- AdisInsight (Adis International Ltd, p.o.S.S.B.M. Drug Profile (GDC-0310). 2016 [cited 2016 Jan] Available from: http://adisinsight.springer.com/drugs/800043573.
- Dainippon Sumitomo Pharma Co., L. Supplementary financial data for the third quarter of the year ending March 31, 2013. [ updated 2013 Jan 31; cited 2016 January]. Available from: http://www.ds-pharma.com/pdf_view.php?id=278.
- Gursahani H, Kim G, Gogas K, et al. NKTR-171: A novel sodium channel blocker for neuropathic pain with reduced CNS side effects. Abstract # 8106/ Poster #JJ11: Presented at the Society for Neuroscience 42nd Annual Meeting: Neuroscience 2012; 2012 Oct 13–17; New Orleans, LA. [cited 2012 Oct 13–17].
- Chromocell Corporation, N.B.T., USA. Analgesia program. 2016 [cited 2016 January]. Available from: http://www.chromocell.com/index2.php?sprache=eng&nav=therapeutics&unternav=therapeutics_programs&intern=therapeutics_programs.
- De Lera Ruiz M, Kraus RL. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J Med Chem. 2015;58(18):7093–7118.
- Wheeler DW, Lee MC, Harrison EK, et al. Case report: neuropathic pain in a patient with congenital insensitivity to pain [version 2; referees: 2 approved]. F1000Research. 2015;3(135).
- Lee JH, Park. CK, Chen G, et al. A monoclonal antibody that targets a Nav1.7 channel voltage sensor for pain and itch relief. Cell. 2014;157(6):1393–1404.
- Dehen H, Willer JC, Prier S, et al. Congenital insensitivity to pain and the “morphine-like” analgesic system. Pain. 1978;5(4):351–358.
- Minett MS, Pereira V, Sikandar S, et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat Commun. 2015;6:8967.
- Minett MS, Eijkelkamp N, Wood JN. Significant determinants of mouse pain behaviour. PLoS One. 2014;9(8):e104458.
- Minett MS, Nassar MA, Clark AK, et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun. 2012;3:791.
- Poras H, Bonnard E, Dangé E, et al. New orally active dual enkephalinase inhibitors (DENKIs) for central and peripheral pain treatment. J Med Chem. 2014;57(13):5748–5763.
- Miyakawa H, Woo SK, Dahl SC, et al. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc Natl Acad Sci U S A. 1999;96(5):2538–2542.
- Akopian AN, Souslova V, England S, et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2(6):541–8.
- Nassar MA, Levato A, Stirling LC, et al. Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8. Molecular Pain. 2005;1:24.
- Zimmermann K, Leffler A, Babes A, et al. Sensory neuron sodium channel Nav1. 8 is essential for pain at low temperatures. Nature. 2007;447(7146):856–859.
- Bierhaus A, Fleming T, Stoyanov S, et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med. 2012;18(6):926–933.
- Faber CG, Lauria G, Merkies IS, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci U S A. 2012;109(47):19444–19449.
- Huang J, Yang Y, Zhao P, et al. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J Neurosci. 2013;33(35):14087–14097.
- Han C, Vasylyev D, Macala LJ, et al. The G1662S Nav1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. J Neurol Neurosurg Psychiatry. 2014;85(5):499–505.
- Chambers JC, Zhao J, Terracciano CM, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42(2):149–152.
- Bagal SK, Bungay PJ, Dentom SM, et al. Discovery and optimization of selective Nav1.8 modulator series that demonstrate efficacy in preclinical models of pain. ACS Med Chem Lett. 2015;6(6):650–654.
- Verkerk AO, Remme CA, Schumacher CA, et al. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res. 2012;111(3):333–343.
- Holm H, Gudbjartsson DF, Arnar DO, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42(2):117–122.
- Sotoodehnia N, Isaacs A, De Bakker PI, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42(12):1068–1076.
- Pfeufer A, Van Noord C, Marciante KD, et al. Genome-wide association study of PR interval. Nat Genet. 2010;42(2):153–159.
- Yang T, Atack TC, Stroud DM, et al. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111(3):322–332.
- Lolignier S, Amsalem M, Maingret F, et al. Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS One. 2011;6(8):e23083.
- Leo S, D’Hooge R, Meert T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav Brain Res. 2010;208(1):149–157.
- Priest BT, Murphy BA, Lindia JA, et al. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel Nav1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci U S A. 2005;102(26):9382–9387.
- Amaya F, Wang H, Costigan M, et al. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26(50):12852–12860.
- Hillsley K, Lim JH, Stanisz A, et al. Dissecting the role of sodium currents in visceral sensory neurons in a model of chronic hyperexcitability using Nav1.8 and Nav1.9 null mice. J Physiol. 2006;576(Pt 1):257–267.
- Sleeper AA, Cummins TR, Dib-Hajj SD, et al. Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J Neurosci. 2000;20(19):7279–7289.
- Luiz AP, Kopach O, Santana-Varela S, et al. The role of Na1.9 channel in the development of neuropathic orofacial pain associated with trigeminal neuralgia. Mol Pain. 2015;11(1):72.
- Huang J, Han C, Estacion M, et al. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain. 2014;137(Pt 6):1627–1642.
- Zhang XY, Wen J, Yang W, et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet. 2013;93(5):957–966.
- Han C, Yang Y, De Greef BT, et al. The domain II S4-S5 linker in Nav1.9: a missense mutation enhances activation, impairs fast inactivation, and produces human painful neuropathy. Neuromolecular Med. 2015;17(2):158–169.
- Leipold E, Leibmann L, Korenke GC, et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat Genet. 2013;45(11):1399–1404.
- Brackenbury WJ. Voltage-gated sodium channels and metastatic disease. Channels (Austin). 2012;6(5):352–361.
- Jansson KH, Castillo DG, Morris JW, et al. Identification of beta-2 as a key cell adhesion molecule in PCa cell neurotropic behavior: a novel ex vivo and biophysical approach. PLoS One. 2014;9(6):e98408.
- Brisson L, Gillet L, Calaghan S, et al. Na(V)1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H(+) efflux in caveolae. Oncogene. 2011;30(17):2070–2076.
- Brisson L, Reshkin SJ, Gore J, et al. pH regulators in invadosomal functioning: proton delivery for matrix tasting. Eur J Cell Biol. 2012;91(11–12):847–860.
- Morinville A, Fundin B, Meury L, et al. Distribution of the voltage-gated sodium channel Na(v)1.7 in the rat: expression in the autonomic and endocrine systems. J Comp Neurol. 2007;504(6):680–689.
- Dib-Hajj SD, Yang Y, Black JA, et al. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14(1):49–62.
- Black JA, Hoeijmakers JG, Faber CG, et al. Nav1.7: stress-induced changes in immunoreactivity within magnocellular neurosecretory neurons of the supraoptic nucleus. Mol Pain. 2013;9:39.
- Weiss J, Pyrski M, Jacobi E, et al. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature. 2011;472(7342):186–190.
- Salat K, Kowalczyk P, Gryzio B, et al. New investigational drugs for the treatment of neuropathic pain. Expert Opin Investig Drugs. 2014;23(8):1093–1104.
- ClinicalTrial.gov (A service of the U.S. National Institutes of Health). Safety & efficacy study of subcutaneous tetrodotoxin for moderate to severe inadequately controlled cancer-related pain (TEC-006). [ updated 2016 Jan 04; cited 2016 Jan]. Available from: https://www.clinicaltrials.gov/ct2/show/nct00725114.
- Le Guiner C, Stieger K, Toromanoff A, et al. Transgene regulation using the tetracycline-inducible TetR-KRAB system after AAV-mediated gene transfer in rodents and nonhuman primates. PLoS One. 2014;9(9).
- Katzel D, Nicholson E, Schorge S, et al. Chemical-genetic attenuation of focal neocortical seizures. Nat Commun. 2014;5:3847.
- Koenig J, Werdehausen R, Linley JE, et al. Regulation of Nav1.7: a conserved SCN9A natural antisense transcript expressed in dorsal root ganglia. PLoS One. 2015;10(6):e0128830.