
Abstract
Objective: The present work evaluated whether the prepared nanoparticles (NPs) would be able to target the drug to the brain by a non-invasive nasal route enhancing its bioavailability.
Methods: Bromocriptine (BRC) chitosan NPs (CS NPs) were prepared by ionic gelation method. The biodistribution, pharmacokinetic parameters and dopamine concentration was analysed by ultra-HPLC/mass spectrometry method. The histopathological examination in haloperidol-induced Parkinson's disease in mice model following intranasal (i.n.) administration was evaluated.
Results: BRC was found stable in all exposed conditions and the percentage accuracy observed for intra-day and inter-day batch samples ranged from 90.5 to 107% and 95.3 to 98.9% for plasma and brain homogenates, respectively. BRC-loaded CS NPs showed greater retention into the nostrils (42 ± 8.5% radioactivity) for about 4 h, whereas the 44 ± 7.5% could be retained up to 1 h for BRC solution. The brain:blood ratios of 0.96 ± 0.05 > 0.73 ± 0.15 > 0.25 ± 0.05 of BRC-loaded CS NPs (i.n.) > BRC solution (i.n.) > BRC-loaded CS NPs (intravenous), respectively, at 0.5 h indicated direct nose-to-brain transport bypassing blood–brain barrier. BRC-loaded CS NPs administered intranasally showed significantly high dopamine concentration (20.65 ± 1.08 ng/ml) as compared to haloperidol-treated mice (10.94 ± 2.16 ng/ml) (p < 0.05). Histopathology of brain sections showed selective degeneration of the dopaminergic neurons in haloperidol-treated mice which was markedly reverted by BRC-loaded CS NPs.
Conclusion: Nanoparticulate drug delivery system could be potentially used as a nose-to-brain drug delivery carrier for the treatment of Parkinson's disease.
1. Introduction
Parkinson's disease (PD) is a neurodegenerative disorder characterised by a progressive loss of dopaminergic neurons in substantia nigra. It is well known that the disabling symptoms in PD are primarily due to degeneration of dopaminergic neurons which cause loss of dopamine (DA) input to striatum from substantia nigra Citation[1,2]. PD is the most common neurodegenerative disorder in the USA today, affecting approximately 1 million individuals Citation[3,4]. In India, PD is more prevalent in rural (41/105) than urban (14/105) population and is more common among men Citation[3]. There is no cure for PD, since currently available therapies can neither arrest nor reverse the progression of the disease. However, the symptoms can be managed with several different drugs. Current therapies for PD ameliorate symptoms in the early phases of disease but become less effective over time Citation[5]. Various categories of antiparkinsonian drugs, which are most often prescribed by physicians, and the ones that are most carelessly taken by the patients have several side effects due to ineffective brain targeting. Due to non-targeting issues, high dose of drug is needed and frequent administration is required which makes the patients noncompliant. Not much notice has been given to the development of a drug delivery system which could deal with the targeting capability of antiparkinsonian agent for brain. The blood-brain barrier (BBB) impedes the brain drug delivery in patients who suffer from PD. Although some drugs exhibit high therapeutic efficacy in vitro, their safe and effective delivery into the brain remains a challenge. In the past few years, intranasal (i.n.) delivery has come to the forefront as an alternative to invasive delivery methods to bypass the BBB and target drugs directly to the brain. This may provide significant advantages such as non-invasiveness, bypassing BBB and decreasing systemic drug exposition, thereby reducing systemic side effects as compared to current conventional strategies Citation[6,7]. The drug may be transported directly into the brain tissue (i.e., olfactory bulb) or the cerebrospinal fluid through the olfactory region of the nasal cavity Citation[7,8]. The strategy to achieve improved delivery to the brain would be a combination of a system that specifically targets the olfactory region and bioadhesive characteristic that retains the formulation at the absorption site for prolonged period of time and improves drug delivery Citation[9]. Nasal mucoadhesive nanoparticles (NPs) can be explored to target drugs to the brain, providing more opportunities for antiparkinsonian agent to enter the brain, thereby reducing its side effects, dose and frequency of dosing Citation[10,11]. Among several NPs prepared from biodegradable polymers, NPs based on chitosan (CS) are desirable as CS is a linear cationic polysaccharide and has been widely considered as a versatile polymer for development of delivery systems due to its biological properties such as biocompatibility, biodegradability, low toxicity, high loading and good delivery ability for hydrophilic molecules Citation[12]. CS has a special feature of being able to adhere to nasal mucosal surface for prolonged periods of time due to mucoadhesive property and improve drug absorption by delaying mucociliary clearance. It also transiently opens tight junctions between epithelial cells and enhances absorption, due to its positive surface charge Citation[13]. Bromocriptine (BRC), an ergot derivative with dopamine receptor agonist activity, has been widely used clinically to delay and minimise the deleterious motor fluctuations associated with long-term levodopa treatment in PD. In addition to its action as a dopamine receptor agonist, BRC is known to directly and indirectly protect dopaminergic cells. BRC has also been reported to have antioxidant effects, inhibit free radical formation and scavenge free radicals and retard apoptosis, which is probably accelerated in PD Citation[14,15]. BRC is available in tablet and capsule dosage form (2.5 – 20 mg/day, in divided doses), and these dosage forms exhibit low bioavailability (6%) due to extensive first-pass metabolism and non-targeted delivery results in side effects such as GI side effects, headache and dizziness Citation[16]. The long-acting injectable form, transdermal and vaginal delivery route of BRC have been already reported Citation[17].
Several analytical procedures for quantifying the levels of BRC have been published Citation[18]. HPLC methods have been reported for the quantification of BRC in pharmaceutical formulation and biological fluids. HPLC methods showed low sensitivity and required extended chromatography run times in order to be specific against other co-extracted plasma compounds Citation[18]. Radioimmunoassay provided good sensitivity but was not sufficiently selective, due to extensive metabolism of BRC Citation[19]. Gas chromatography revealed a higher sensitivity but required a complex extraction procedure. Mass spectrometry coupled to liquid chromatography (LC-MS) has been extensively used for BRC determination in plasma (pg/ml) range Citation[20], since rapid quantification of BRC in nanogram ranges was not extensively determined in brain homogenates with LC–MS. The literature survey revealed that till date stability indicating assay method has not been reported. In the present study, a standard hyphenated technology as ultra HPLC/electron spray ionisation/quadrupole-time of flight-mass spectrometry (UHPLC/ESI-q-TOF-MS) method was used, as it utilised high linear velocities which dramatically improved peak resolution, sensitivity and speed of analysis Citation[21].
The present work investigates the development and validation of stability indicating ultra-HPLC/mass spectrometry (UHPLC/MS) in biological fluids. The present work also evaluates the ability of BRC-loaded CS NPs to be distributed to brain regions and plasma, together with the estimation of dopamine and pharmacokinetic parameters of BRC-loaded CS NPs by UHPLC/MS method. The stability study and shelf life of optimised formulation according to International Conference of Harmonization of Technical Requirements for registration of Pharmaceuticals for Human use (ICH) guidelines was also evaluated. Moreover, the present work also evaluates whether the prepared NPs would be able to target the drug to the brain by non-invasive nasal route, besides enhancing its bioavailability.
2. Materials and methods
CS with medium molecular weight and degree of deacetylation about 85% and sodium tripolyphosphate (TPP) were purchased from Sigma-Aldrich, St. Louis, MO, USA. Stannous chloride dihydrate (SnCl2·2H2O) was purchased from Sigma-Aldrich, St. Louis, MO, USA. Sodium pertechnetate, separated from molybdenum-99 (99m) by solvent extraction method, was provided by Regional Center for Radiopharmaceutical Division (Northern Region), Board of Radiation and Isotope Technology, New Delhi, India. Instant thin layer chromatography silica gel strips were purchased from Gelman Science, Inc., Ann Arbor, MI, USA. All other reagents and chemicals used were of analytical reagent grade without further purification.
2.1 Preparation and characterisation of CS NPs
BRC-loaded CS NPs were prepared according to ionic gelation method as described in previous report Citation[22]. CS was first dissolved in acetic acid (2% v/v, pH 3.5) at a concentration of 0.175% w/v with constant stirring at 300 rpm overnight. The drug (BRC) at the concentration of 0.5 mg/ml was added to CS solution with constant stirring prior to addition of TPP solution. NPs were obtained by the addition of 3 ml of TPP solution (0.175% w/v) to 10 ml of BRC-mixed CS solution with constant stirring at 600 rpm at room temperature for 30 min. The final CS:TPP mass ratio was equal to 3.3:1 (w/w). The placebo CS NPs were prepared by the same procedure without using BRC. The resultant NPs were concentrated by ultra-centrifugation at 10,000 × g at 4°C for 40 min. The supernatant was used to determine entrapment and drug loading (DL) and the pellets were washed with distilled water and then freeze-dried for further characterisation study.
Particle size and polydispersity index (PDI) of all optimised NPs were determined by photon correlation spectroscopy (PCS) using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK). Each sample was suitably diluted with filtered distilled water to avoid multi-scattering phenomena and was placed in a disposable sizing cuvette. The PDI was studied to determine the homogeneity of the particle size distribution. The size analysis of a sample consisted of three measurements, and the results were expressed as mean size ± SD. The zeta potential is a very useful parameter for evaluating the stability of any colloidal system. The determination of the zeta potential was performed by Laser Doppler Anemometry (Zetasizer Nano ZS Malvern Instruments, Worcestershire, UK). The particles size of the prepared NPs was also determined by using transmission electron microscopy (Morgagni 268D TEM, MA, USA). The surface morphology of the prepared NPs was carried out by using scanning electron microscopy (SEM) techniques (EVO LS 10 Zeiss, Carl Zeiss, Inc., Germany).
2.2 Determination of entrapment efficiency, DL and in vitro release study
The entrapment efficiency (EE) and DL of CS NPs were determined by the separation of CS NPs in the form of pellets from the acidified solution of water (2% v/v acetic acid, pH 3.5) containing free drug by ultra-centrifugation at 10,000 × g at 4°C for 40 min. The amount of free BRC in the supernatant was measured spectrophotometrically at 305 nm Citation[23]. Dilutions of samples and calibration curves were prepared in acidified solution of water (2% v/v acetic acid, pH 3.5). The % EE and % DL were calculated using the following equations:
In vitro release of BRC from CS NPs was evaluated by the dialysis bag diffusion technique reported by Md et al. Citation[22,24].
2.3 Nasal clearance study
The deposition and subsequent clearance of the drug from the mice nose was investigated by gamma scintigraphy study. Swiss albino mice of either sex weighing between 20 and 40 g were selected for the study. Three mice for each formulation per time point were used in the study. Radiolabeled 99mTc-pertechnetate BRC solution and BRC-loaded CS NPs were prepared by adding 0.5 ml BRC formulations in sterile glass vial followed by addition of concentrations of a reducing agent, stannous chloride (60 and 120 µg in 0.1 M acetic acid, respectively). The resultant solutions were incubated for 10 min at room temperature and pH was adjusted to 5.5 and 6.5 using 50 mM sodium bicarbonate solution, respectively, for both formulations. The resultant mixture is the so-called reduced technetium. To the resultant solutions the required volume of 99mTc-pertechnetate (1 – 2 mCi) was added with continuous mixing and the solution was incubated at 25 ± 5°C for 30 min. Then the radiolabeled complex of 99mTc-BRC solution and 99mTc-BRC-CS NPs solution (100 µCi/10 µl) containing BRC (equivalent to 0.22 mg/kg body weight) was administered (5 µl) in each nostril. Formulations were infused into the nostrils with the help of low-density polyethylene (LDPE)-made tip put on the micropipette (20 µl) at the delivery site for i.n. administration. Immediately after administration and at different time points the animals were laid on their backs under the monitor of single photon emission computerised tomography (SPECT, LC 75-005, Diacam, Siemens AG, Erlanger, Germany) gamma camera and nasal clearance of formulations were observed. The mice were anaesthetised by intramuscularly injecting 0.2 ml ketamine (50 mg/ml) and the mice were placed on the imaging board. Regions of interest were drawn around the site of deposition and the total activity in the area was determined. Percentage activity remaining in nose was calculated at different time points with respect to the initial activity and was plotted against time Citation[25].
2.4 UHPLC/MS analysis and chromatographic condition
The UHPLC/MS method was used for the quantification of BRC in plasma and brain homogenates. A UHPLC/MS method was developed and validated for the determination of BRC in biological samples. The method was validated in accordance with current acceptance criteria of ICH guidelines Citation[26]. The system consisted of UHPLC coupled with mass spectrometer system (Waters ACQUITY UPLC™, Waters Corp., USA). BEH C18, 100.0 × 2.1 mm; 1.7 m (ACQUITY UPLC™, Waters Corp., MA, USA) column was used as stationary phase. The mobile phase for UHPLC analysis consisted of acetonitrile: ammonium acetate (2 mM) (70:30, v/v), which was degassed prior to use. The total chromatographic run time was 6.0 min. The flow rate was set at 0.25 ml/min and 10 µl of sample solution was injected in each run. Quantitation was performed using Synapt Mass Spectrometery (Synapt MS) of the transitions of m/z 656 → 348 for BRC with a scan time of 1.0 min and 0.02 s interscan per transition. The optimal MS parameters were as follows: capillary voltage 3.0 kV, cone voltage 20 V, source temperature 120°C and desolvation temperature 350°C. The optimum values for compound-dependent parameters like trap collision energy was set to 26.3 V, respectively, for fragmentation information. Nitrogen was used as the desolvation gas with a flow rate of 500 L/h. Argon was used as the collision gas at a pressure of approximately 5.3 × 10−5 torr. The accurate mass and composition for the precursor ions and for the fragment ions were collected in multi-channel analysis mode and were acquired and processed using MassLynx V 4.1 software Citation[20].
2.4.1 Preparation of standard working solutions
A standard stock solution of BRC and internal standard (IS) (propranolol) containing a 100 µg/ml solution was prepared separately in methanol. All subsequent dilutions were made in methanol. Standard working solution of BRC at concentration of 5, 10, 20, 40, 80, 160 and 200 ng/ml was prepared. The IS stock solution was diluted to a working concentration of 10 ng/ml. These working solutions were stored at +4°C until use.
Plasma and brain homogenate standards of BRC were prepared fresh daily by spiking 50 µl of the appropriate working solutions into 1 ml of heparinised plasma and brain homogenate to yield calibration concentrations of 0.25, 0.5, 1, 2, 4, 8 and 10 ng/ml. Methanol (50 µl) was added to control plasma and brain homogenate to make up plasma and brain homogenates blank samples, respectively. Plasma and brain homogenates quality control (QC) samples were prepared by adding appropriate QC working solutions and plasma and brain homogenate volumes to yield concentrations of 0.5, 4 and 8 ng/ml. After preparation, the bulk QC samples were aliquoted into 2.5 ml volumes and stored at -25°C until used.
2.4.2 Samples extraction procedure
A 1 ml aliquot of plasma and brain homogenates was spiked with 200 µl of orthophosphoric acid solution (5% in water), followed by 100 µl of IS working solution separately. Orthophosphoric acid solution was added as a protein-precipitating agent. The sample was vortexed for 10 min in a shaker. After vortexing, 3 ml of ethyl acetate was added to the solution, then vortexed again and the sample was kept on an ice bath for 5 min. The resultant mixture was then centrifuged (Centrifuge, Eppendrof Model 5810R, Eppendrof, Germany) at (1000 × g; 10 min). The supernatant (2 ml) was transferred to borosilicate glass tubes and evaporated to dryness with nitrogen evaporator at 25 ± 0.5°C and 15 psi. About 500 μl of reconstitution solution (acetonitrile–2 mM ammonium acetate (70:30 v/v) was then added and vortexed for 10 s. The solution was transferred into the clean autosampler vials and 10 μl was programmed to be injected into UHPLC/MS system for analysis.
2.4.3 Method validation
The linearity of the method was determined by analysing three standard plots containing seven non-zero concentrations. Peak area of analyte was utilised for the construction of calibration curves, using linear least squares regression of the plasma concentrations and the measured peak area. All analyses were performed in three replicates (n = 3). QC samples (theoretical contents) were applied at three different levels as low (LQC: 0.5 ng/ml), medium (MQC: 4 ng/ml) and high (HQC: 8 ng/ml). For determining the intra-day accuracy and precision, replicate analysis of biological samples of BRC was performed on the same day. The inter-day accuracy and precision were assessed by analysing three precision and accuracy batches on three consecutive validation days followed by comparing them with the calibration curves prepared on the same day and on three different days. Precision was expressed as the percentage relative to the standard deviation of measured concentrations for each calibration level, whereas accuracy was expressed as percentage recovery (determined concentration/nominal concentration ×100) of drug added to the blank biological samples. Sensitivity of the method was proved by establishing the limit of detection (LOD) and limit of quantification (LOQ) for BRC with a signal-to-noise ratio of 3:1 and 10:1, respectively. LOD and LOQ were determined by injecting a series of diluted solutions having known concentrations of BRC spiked with plasma and brain homogenates samples. The stability study was conducted as per ICH guidelines Citation[27], according to which a compound is deemed ‘stable' if the concentration of the compound through the stability experiment does not deviate by > 15% compared to a control (time 0) concentration. BRC-spiked plasma and brain homogenates of QC sample concentrations at 4 – 8 ng/ml were analysed as fresh preparations and were then left in the autosampler at room temperature for 24 h (bench top stability) and at −80°C for 30 days (long-term stability) to investigate the processed sample stability.
2.4.4 Forced degradation study
Forced degradation studies were performed using the following conditions: acid hydrolysis (2 N HCl at 100°C for 2 h), alkaline hydrolysis (2 N NaOH at 100°C for 2 h), oxidation (30% H2O2 at 100°C for 2 h), photolytic (sunlight for 8 h) and thermal hydrolysis (80°C for 72 h) (ICH, 2005). All forced degradation samples were analysed at concentration of 4 ng/ml. In all degradation studies, percentage recoveries of the drug and degradation products after triplicate application were calculated.
2.5 In vivo studies
Swiss albino mice of either sex weighing between 20 and 40 g were selected for the in vivo studies. The animals were housed two per cage at 20 – 25°C with free access to food and water with a 12-h light–dark cycle. All research and testing activities related to this manuscript were reviewed and approved by the Institutional Animal Ethical Committee of Jamia Hamdard, New Delhi, India (protocol No: 853) before the initiation of the study and during its execution.
2.5.1 Brain and blood distribution of BRC-loaded CS NPs
Three mice for each formulation per time point were used in the study. BRC-loaded CS NPs solution (group I) containing BRC (equivalent to 0.22 mg/kg body weight) was injected through the tail vein of mice. The BRC solution (group II) and BRC-loaded CS NPs solution (group III) containing BRC (equivalent to 0.22 mg/kg body weight) was administered (5 µl) in each nostril. Formulations were infused into the nostrils with the help of LDPE-made tip put on the micropipette (20 µl) at the delivery site for i.n. administration. The i.n. dose was administered to mice without any anaesthesia or sedation. The mice were held from the back in slanted position and were placed in supine position with head angle at 0° (horizontal) during administration as well as were held for approximately 2 min after administration of doses. The mice were anaesthetised with 10% chloral hydrate solution and sacrificed humanely by cervical dislocation method at different time intervals (0.5, 1, 2, 4 and 8 h) and the blood was collected using cardiac puncture Citation[28]. The collected blood was mixed properly with the anticoagulant and centrifuged at 1000 × g for 10 min. The plasma was separated and stored at −21°C until drug analysis was carried out using UHPLC/MS (Acquity UPLC™ system, Waters, Miliford, CT, USA). At the same interval of blood collection, brain tissue was also taken out by decapitation method and washed by physiological saline solution. Whole brain tissue was homogenised using a laboratory homogeniser with physiological saline and then centrifuged at 1000 × g for 10 min, and aliquots of the supernatant were separated and stored at −21°C until drug analysis was carried out using UHPLC/MS.
2.5.2 Pharmacokinetic studies
Pharmacokinetic parameters for formulations were calculated by pharmacokinetic software (PK Functions for Microsoft Excel, Pharsight Corp., Mountain view, CA, USA). The Cmax of BRC and the T max were obtained directly from the actual plasma profiles. The degree of BRC targeting to brain after i.n. administration was determined by the drug targeting index which can be described as the ratio of the value of AUCbrain:AUCblood following i.n. and intravenous (i.v.) administration Citation[29].
2.5.3 Brain targeting studies
Brain targeting efficiency, that is, drug targeting efficiency (%DTE) that represents time average partitioning ratio was calculated as follows:
Nose to brain direct transport percentage (%DTP) was calculated as follows:
The percentage absolute bioavailability value (F, %) of the i.n. formulations was calculated from equation
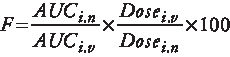
where Bx = (Bi.v./Pi.v.) × Pi.n., Bx is the brain AUC fraction contributed by systemic circulation through the BBB following i.n. administration. Bi.v. is the AUC0 – α (brain) following i.v. administration. Pi.v. is the AUC0 – α (blood) following i.v. administration. Bi.n. is the AUC0 – α (brain) following i.n. administration. Pi.n. is the AUC0 – α (blood) following i.n. administration Citation[28].
2.5.4 Estimation of dopamine
Dopamine was estimated by LC-MS (4000 QTRAP LC-MS/MS System, AB Sciex, Foster city, CA, USA) with a hybrid triple quadrupole/linear ion trap mass spectrometer equipped with pressure ion (PI) method, which was already developed and validated by Fenli et al. Citation[30]. The analytes were separated on a thermo C18 column (4.6 mm 9 250 mm, 5 lm, SN: 1245575T, Thermo Electron Corp., USA) with ambient column temperature. The mobile phase was 0.05% formic acid and acetonitrile (92: 8, v/v) for the determination of dopamine. The flow rate was 0.8 ml/min and the post-column splitting ratio was 4:1. Dopamine was obtained by PI in the positive ion mode. Calibration curve of standard dopamine (Sigma Aldrich, St. Louis, MO, USA) were prepared in deionised water with seven calibration concentrations in the range of 4 – 4100 ng/ml. Then the Swiss albino mice were decapitated and brains were removed and kept on ice immediately, within 4 min and striatum were removed. Striatum (50 mg) was homogenised in 500 µl ice-cold methanol by vortex mixing for 10 min. About 1ml of acetonitrile was mixed with homogenate in 2.5 ml conical plastic centrifuge tube and centrifuged at 1000 × g for 20 min. Then the supernatant was evaporated to dryness by nitrogen evaporator at 14°C. The dry residue was then reconstituted with 400 μl mobile phase consisting of 0.05% formic acid and acetonitrile (92: 8 v/v). The samples were vortexed for 10 s then the mixture was again centrifuged at 1000 × g for another 20 min. The upper aqueous layer was injected into the LC-MS/MS for analysis of dopamine.
2.5.5 Histopathological study
At the end of the behavioural studies, hematoxylin and eosin staining was used for examining the histology of brain with light microscopy. Histological evaluation was performed on formalin-fixed paraffin-embedded sections. It was performed in order to assess whether or not the administration of haloperidol caused neuronal damage and whether treatment with BRC-loaded CS NPs improved neuronal damage. Normal brain (negative control), brain treated with haloperidol (positive control), brain treated with BRC-loaded CS NPs were fixed immediately in 10% formalin and embedded in paraffin after isolation. After that 5 µm thick sections were cut and deparafinised using xylene and ethanol. The sections were stained with hematoxylin and eosin. The stained sections were then observed under the microscope at 40× magnifications Citation[2].
2.6 Stability studies as per ICH guidelines
As per the ICH guidelines, for the products intended to be stored in a refrigerator, the storage conditions of temperature and relative humidity (RH) for accelerated stability studies are 25 ± 2°C and 60 ± 5% RH Citation[27]. Three batches of each of the BRC-loaded CS NPs suspensions were prepared. These nanosuspensions were kept at a temperature of 25 ± 2°C/ 60 ± 5% RH for 6 months. Samples were withdrawn after specified time intervals (0, 1, 2, 3 and 6 months) and physical appearance, particle size, PDI and drug content of the samples were determined. For the determination of shelf life by conventional method, nanosuspension was kept at 25 ± 2°C; 30 ± 2°C and 35 ± 2°C and 60 ± 5% RH for 3 months. Samples were withdrawn after specified time intervals (0, 30, 60 and 90 days) and the remaining drug content was determined by UHPLC/MS method. Logarithm of percentage drug remaining versus time (in days) was plotted. The degradation rate constant ‘k' was determined from the slope of the lines at each elevated temperature using the given equation Slope = −k/2.303. Plot of the logarithm of k values at various elevated temperatures against the reciprocal of absolute temperature was drawn (Arrhenius plot). From the plot, k value at 5°C was determined and was used to calculate shelf life by substituting in the equation t0.9 = 0.1054/ k5, where, t0.9 is the time required for 10% degradation of the drug and is referred to as shelf life.
2.7 Statistical analysis
All data were expressed as mean ± SD. For multiple-group comparison, one-way analysis of variance was used followed by Bonferroni tests. Comparison between all groups versus control was carried out with a Dunnett's t-test. Differences were considered statistically significant at p < 0.05.
3. Results and discussion
3.1 Preparation and characterisation of NPs
The preparation, optimisation and physical characterisations of BRC-loaded CS NPs have been already reported in our earlier publication Citation[22]. The prepared NPs exhibited an average particle size < 200 nm under TEM, which was in good agreement with the hydrodynamic-based particle size measured by the PCS having particle size of 161.3 ± 4.7 nm. The prepared NPs showed acceptable PDI of 0.44 ± 0.03. The EE and LC of BRC-loaded CS NPs were 84.2 ± 3.5 and 37.8 ± 1.8%, respectively. The mean zeta potential of BRC-loaded CS NPs was found to be +40.32 ± 2.78 mV, which revealed that prepared formulations were stable Citation[31]. The surface morphology of CS NPs recorded from SEM was either spherical or ellipsoidal with smooth surface (figure not shown) Citation[15]. The percentage release profile of BRC from the nanoparticulate formulation was investigated in phosphate buffer solution (pH 7.4). The release profile of BRC from the nanoparticulate formulation showed two-step biphasic process, that is, an initial burst effect (30.98 ± 1.56%) for 1 h followed by subsequent slower release (87.80 ± 2.43%) for 24 h showing a sustained release of the drug from the formulation Citation[22].
3.2 Nasal clearance study
BRC-loaded CS NPs showed greater ability to retain in the nostrils with retention of about 42 ± 8.5% radioactivity for about 4 h, whereas the 44 ± 7.5% could be retained up to 1 h for control formulation (). The decreased clearance of BRC-loaded CS NPs was due to mucoadhesive properties of CS present in the formulation. The positively charged amino groups on CS adhered to the negatively charged sialic acid residue of the mucosa Citation[25].
Figure 1. Percentage radioactivity remaining in the nose after different time intervals for BRC-loaded CS NPs and BRC solution (control) (n = 3, mean ± SD) is shown.
3.3 Method development and validation
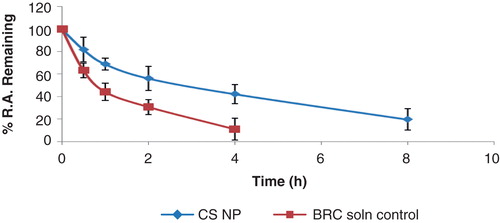
In this study, good separation for BRC was best achieved in a mobile phase consisting of a mixture of acetonitrile (70%) and 2 mM ammonium acetate buffer (30%) at flow rate of 0.25 ml min. The mass spectrometric conditions were optimised to obtain maximum sensitivity. BRC can be ionised under either positive or negative electrospray ionisation conditions. Due to the presence of basic nitrogen in the molecule, BRC exhibited favourable sensitivity in positive ion mode detection because of the efficiency of ionisation of the analyte. The typical mass spectrum of BRC and propranolol as a IS are shown in . The major product ion included m/z 656 and daughter ion MS/MS m/z 348 for BRC and m/z 260 and daughter ion m/z 183 for propranolol. In order to enhance and stabilise the response, ammonium acetate was added in the mobile phase. The liquid–liquid extraction procedures were used to prepare BRC samples in biological samples for the study. The use of Waters ACQUITY UHPLC™ BEH C18 (100 × 2.1 mm; 1.7 μm) column at 40 ± 2°C helped in separating and eluting both analytes, BRC and IS. Selectivity was assessed by analysing plasma and brain homogenates from mice and examining for peaks that interfered with BRC and the IS. Representative chromatograms obtained from plasma spiked with BRC and brain spiked with BRC is shown in . There were no chromatographically interfering peaks observed at the retention times of either analyte or IS in the samples. Under the chromatographic conditions described, the mean retention times of BRC and the IS were 2.95 and 1.85 min, respectively. The limit of quantification for BRC was 0.11 and 0.063 ng/ml, respectively, in plasma and brain homogenates. The limit of detection for plasma and brain homogenates was found to be 0.038 and 0.02 ng/ml, respectively. The calibration curves were constructed with seven concentrations in triplicate ranging from 0.25 to 10 ng/ml for plasma and brain homogenates. The peak area of the drug was plotted against the concentration and the regression equation obtained was y = 6506X + 940 and 5989X + 497 with the correlation coefficient of 0.988 and 0.985 for plasma and brain homogenates, respectively. The results showed excellent correlation between the peak area and concentration of BRC. The extraction recoveries of BRC in plasma were 80.54 ± 2.02, 83.61 ± 1.80 and 82.80 ± 4.25% for QC samples, respectively. The extraction recoveries in brain homogenates were found to be 84.19 ± 2.3, 81.56 ± 2.5 and 80.0 ± 3.8%, respectively, for QC samples. The extraction recovery of IS was 83.67 ± 3.5%. These results indicated that the sample procedure of protein precipitation with orthophosphoric acid (5%) and ethyl acetate was efficient for the extraction of BRC in plasma and brain homogenates. Inter-day and intra-day precision and accuracy of the method were determined by analysis of QC samples containing the three analytes at three different concentrations. The results obtained are recorded in . The percentage accuracy observed for intra-day batch samples ranged from 90.5 to 107% for plasma and 95.3 – 98.9% for brain. However, the percentage accuracy observed for inter-batch samples ranged from 94.2 to 105.9% for plasma and 93.30 to 98.34% for brain, respectively (). The method showed reproducibility with an intra-day and inter-day precision method Citation[20].
Table 1. Validation data of BRC in biological samples.
Figure 3. Typical chromatograms of (A) extracted BRC from plasma; (B) extracted BRC from brain homogenates are shown.
Figure 2. Mass spectrum of BRC precursor ion (protonated precursor [M+H]+ ions at m/z 656.12) and propranolol precursor ion (protonated precursor [M+H]+ ions at m/z 260.10) are shown.
![Figure 2. Mass spectrum of BRC precursor ion (protonated precursor [M+H]+ ions at m/z 656.12) and propranolol precursor ion (protonated precursor [M+H]+ ions at m/z 260.10) are shown.](/cms/asset/d03ba97b-7555-4cfe-8483-b95ac7717641/iedd_a_894504_f0003_b.jpg)
3.4 Stability effect and forced degradation study
The results obtained for short-term and long-term stability study are summarised in . The results indicated that the processed samples were stable in all storage condition. The concentrations found were within the allowed limit ±10% of nominal concentrations, revealing no significant drug loss at room temperature and at −80°C for 30 days. Forced degradation samples under various conditions were analysed at an initial concentration of 4 ng/ml of BRC on UHPLC/MS using PDA detector to ensure homogeneity of BRC peak (). Degradation was not observed when BRC was subjected to acid (2 N HCL at 100°C for 2 h), thermal (80°C for 72 h) and oxidative hydrolysis (30% H2O2 at room temperature for 2 h). The maximum degradations product was observed in photolytic stress condition having 39% degradation with five degradation product, whereas the alkaline hydrolysis showed three degradation products with 36% degradation. All presented results indicated that the method indicated stability Citation[32].
Table 2. Stability data of BRC in biological samples under different conditions.
Figure 4. Degradation products obtained after (A) acidic stress; (B) alkaline stress; (C) oxidation stress; (D) sunlight and (E) thermal stress are shown.
3.5 In vivo study
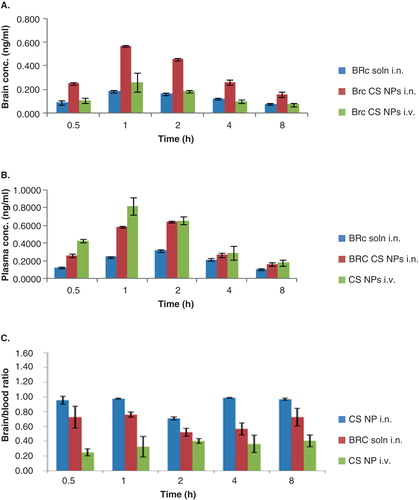
The brain and blood distribution of BRC solution, BRC-loaded CS NPs was quantitatively assessed in mice after i.n. and i.v. administration. As shown in intranasally administered BRC-loaded CS NPs had the highest BRC brain concentration at all time points after dosing. It could be due to faster and better absorption across the nasal mucosa into the olfactory epithelium at 1 h (0.569 ± 0.008 ng/ml), which declined slowly due to sustained release characteristics of the formulation. The BRC solution (Cmax 0.185 ± 0.013 at 1 h) and BRC-loaded CS NPs (Cmax 0.259 ± 0.081 at 1 h) administered intranasally and intravenously showed significantly lesser brain concentration as compared to BRC-loaded CS NPs administered intranasally at all time points (p < 0.05) (). When BRC solution and BRC-loaded CS NPs were administered intranasally, this showed a Cmax of 0.315 ± 0.014 and 0.645 ± 0.011 ng/ml, respectively, in blood at 2 h due to slow absorption across the nasal mucosa into the systemic circulation. BRC-loaded CS NPs increased brain targeting and also increased the absorption in the blood more in comparison to the BRC solution administered intranasally due to fact that the mucoadhesive nature of NPs remained adhered for long period of time to nasal mucosa and CS also acted as a permeation enhancer which may have increased the absorption of drug in blood. However, BRC solution did not remain adhered to nasal mucosa due to lack of mucoadhesive agent and got cleared rapidly due to mucociliary clearance under normal circumstances. When BRC-loaded CS NPs were administered intravenously, the Cmax (0.823 ± 0.099 ng/ml and 0.259 ± 0.081 ng/ml) was reached at 1 h in blood and brain. When BRC-loaded CS NPs was administered intravenously, higher quantity of BRC did not reach in brain because BRC-loaded CS NPs with their high positive surface was rapidly recognised by the reticuloendothelial system in blood Citation[15]. A small quantity of BRC was detected in brain because some of the CS NPs may have crossed through BBB due to small size of NPs and presence of CS, which has also been proven to be an efficient absorption enhancer that can transiently open tight junctions between epithelial cells of BBB and thereby significantly enhance the absorption of BRC in brain Citation[25]. However the blood concentrations of BRC after i.n. administration either as a solution or in the form of NPs, were all significantly lower than those after i.v. administration formulation which can be depicted by p value < 0.05 as shown in . The brain:blood ratio of the drug at all sampling time points for different formulations was determined and is shown in . The brain:blood ratios of 0.96 ± 0.05 > 0.73 ± 0.15 > 0.25 ± 0.05 of BRC-loaded CS NPs (i.n.) > BRC solution (i.n.) > BRC-loaded CS NPs (i.v.), respectively, at 0.5 h are indicative of direct nose-to-brain transport bypassing the BBB, which in turn prove the superiority of nose-to-brain delivery of BRC-loaded CS NPs Citation[29]. Selected pharmacokinetic parameters of i.n. and i.v. administrations calculated from the individual time–concentration profiles from the experiment where values of AUC, Cmax, Kel, half-life and Tmax can be observed are recorded in . Significant differences after i.n. and i.v. administration were observed for Cmax and AUC. When the AUC0 – ∞ of brain concentration of BRC solution (i.n.) (1.992 ± 0.276 ng h/ml) and BRC-loaded CS NPs (i.n.) (3.618 ± 0.37 ng h/ml) were compared, the AUC0 – ∞ of BRC-loaded CS NPs (i.n.) were found to be significantly higher (p < 0.05). The reason could be that the CS acts as a mucoadhesive agent and binds strongly to negatively charged surface of sialic acid present in mucous as a result of which mucin and CS can demonstrate strong electrostatic interaction in solution which significantly decreased the mucociliary clearance; as a result BRC-loaded CS NPs stayed for longer period of time which significantly improved the drug transport from nose to brain Citation[9,11]. The two important parameters %DTE and %DTP representing the percentage of drug directly transported to the brain via the olfactory or trigeminal pathway were calculated. The DTE and DTP values for BRC-loaded CS NPs and BRC solution were 265.6 ± 37.3 and 54.13 ± 11.76% and 192 ± 48.0 and 37.00 ± 13.2%, respectively. The high value of DTE and DTP for BRC-loaded CS NPs as compared to BRC solution revealed a measurable degree of BRC brain targeting Citation[33]. The comparative bioavailabilities of BRC solution and BRC-loaded CS NPs administered intranasally for brain targeting were calculated and statistically analysed. The absolute bioavailability of BRC solution in blood and brain was found to be 77.9 ± 8.27 and 129.5 ± 42.11%, respectively. The absolute bioavailability of BRC-loaded CS NPs in blood and brain was found to be 100.69 ± 4.14 and 231.9 ± 51.20%, respectively. The comparative bioavailability results of BRC solution and BRC-loaded CS NPs revealed that there is a significant enhancement of bioavailability in brain, after administration of the BRC-loaded CS NPs as compared to BRC solution administered intranasally (p < 0.05). This observation could be a substantial achievement of direct nose-to-brain targeting in PD therapy. The increase in bioavailability may be due to protection of encapsulated drug by NPs from harsh environment of nose such as biological and/or chemical degradation, and extracellular transport by P-gp efflux proteins. The other reason could be the avoidance of first-pass metabolism. These reasons would also in turn increase CNS availability of the drug. Thus, the results of the present investigation proved that BRC could be transported directly to the brain after i.n. delivery of CS NPs, thereby enhancing its bioavailability and direct targeting to the brain.
Table 4. Pharmacokinetic parameters of BRC solution and BRC-loaded CS NPs in mice following intravenous (i.v.) and intranasal (i.n.) administration.
Table 3. Brain and blood distribution of BRC solution and BRC-loaded CS NPs in mice after intranasal (i.n.) and intravenous (i.v.) administration.
Figure 5. Concentration–time profile of (A) brain concentration (B) plasma concentration and (C) brain:blood ratio of BRC solution and BRC-loaded CS NPs in mice after intranasal (i.n.) and intravenous (i.v.) administration (n = 3, mean ± SD) is shown.
3.5.1 Dopamine estimation
Effect of haloperidol on dopamine concentration in mice brain striatum was studied and dopamine levels were determined. Dopamine content in saline-treated mice was 21.87 ± 1.04 ng/ml. Haloperidol-treated mice (2.0 mg/kg) showed highly significant decrease (10.94 ± 2.16 ng/ml) in dopamine level as compared to saline-treated mice (p < 0.05). Dopamine exerted a direct excitatory influence on subthalamic nucleus (STN) neurons via activation of D2-like receptors; the excitatory effect of dopamine was reduced by D2 antagonist haloperidol Citation[34]. Small decrease in dopamine content was found in mice co-treated with BRC solution administered either orally or i.n., having concentrations of 14.00 ± 0.37 and 15.71 ± 1.42 ng/ml, respectively, which showed non-significant result among each other (p > 0.05) but relatively significant result as compared to saline (p < 0.05). However, BRC-loaded CS NPs-treated mice showed significantly increased dopamine level (20.65 ± 1.08 ng/ml) as compared to haloperidol-treated mice (p < 0.05). The study showed that dopamine content was significantly higher in BRC-loaded CS NPs-treated mice.
3.5.2 Histopathological studies on brain
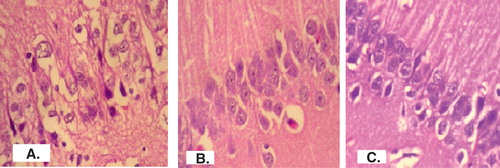
The images of brain tissues were observed for any changes due to haloperidol induced in mice and effect of BRC-loaded CS NPs. The obtained images of histopathological studies in brain are shown in . The high-power photomicrograph of brain from normal mice revealed a portion of the cornu ammonis (CA1) region. All the neurons have large round vesicular nuclei with prominent nucleoli and amphophilic cytoplasm. Neuronal density is 24/high power field (HPF) (). The high-power photomicrograph of brain from mice treated with haloperidol showed a portion of the CA1 region. There was pronounced cytoplasmic vacuolation and shrinkage of nuclei . Neuronal density was reduced to 16/HPF (). High power photomicrograph of brain from mice treated with haloperidol plus BRC loaded CS NPs showing a portion of the CA1 region and most of the neurons structure was similar to brain of normal mice. A few vacuolated neurons are seen along the lower edge. Neuronal density is 23/HPF (). Brain of haloperidol-treated and haloperidol plus BRC-loaded CS NPs-treated mice showed the presence of eosinophilic lesions. These lesions were more in number and bigger in size in the haloperidol-treated mice when compared to co-treated group where the lesions were less in number, small in size and dispersed Citation[35].
Figure 6. Brain section of (A) normal mice, (B) haloperidol-treated mice and (C) combined treatment of haloperidol and BRC-loaded CS NPs (i.n.)-treated mice is shown.
3.6 Stability studies of optimised BRC-loaded CS NP formulation as per ICH guidelines
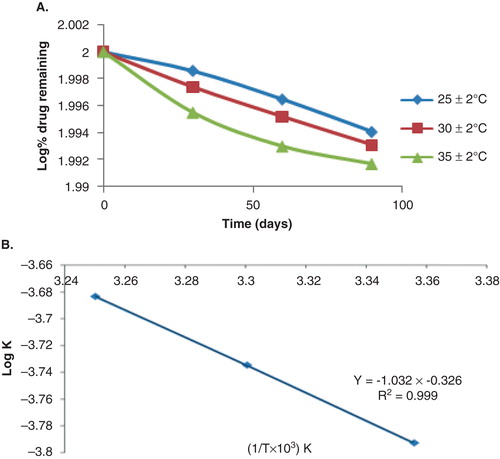
In various literatures it has been shown that, at higher temperature such as 40 ± 2°C, there was little or no CS NPs remaining after 6 months Citation[36]. This may be due to the decrease in molar mass of the CS possibly due to hydrolysis causing scission of the polymer chains, which results in a decrease in NP size and eventual disintegration Citation[36]. So in the present study the developed nanosuspension are intended for storage in a refrigerator. As per the ICH guidelines, for the products intended to be stored in a refrigerator, the storage conditions of temperature and RH for accelerated stability studies are 25 ± 2°C and 60 ± 5% RH. The physical stability of nanosuspension in terms of particle size, PDI and drug content was evaluated. Stability of a drug in a dosage form at different environmental conditions is important, because it determines the shelf life of the formulation. Changes in the physical appearance, colour, odour or taste of the formulation indicate the drug instability. There were no changes in their physical appearance of formulation (opalescent appearance at all time points). At 25°C, the particle size, PDI and drug content (164.6 ± 4.45, 0.518 ± 0.04 nm and 98.01%, respectively) of NPs after 6 months remained similar to those of the freshly prepared samples (161.1 ± 3.65 and 0.445 ± 0.01 nm and 100%, respectively). The changes in the observed parameters were not found to be statistically significant (p > 0.05), which indicated that the optimised formulations were stable. The result at 25°C was consistent with the findings reported by Tsai et al. Citation[37] for NPs of a similar size range. The accelerated stability studies were determined by the percentage drug remaining in the optimised nanosuspension when stored for 3 months at elevated temperatures of 25 ± 2°C; 30 ± 2°C; 35 ± 2°C at 65 ± 5% RH. The order of degradation of BRC in the NPs was determined and was found to follow first-order kinetics as shown in . The reaction rate constant ‘k' for the degradation was measured. Plot of the logarithm of k values for the NPs at each elevated temperature against the reciprocal of absolute temperature was drawn (Arrhenius plot) as shown in . From the plot, k value at 5°C (refrigerator temperature) shelf life of the nanoparticulate formulation was calculated to be 1.44 years.
Figure 7. Representation of (A) log percentage drug remaining versus time and (B) Arrhenius plot for optimised BRC-loaded CS NPs.
4. Conclusion
In the present study, an UHPLC/MS analytical method was established for determining BRC and its nanoparticulate formulation in brain and plasma. Higher brain uptake of nanoparticulate formulations after i.n. administration could be due to both the olfactory and trigeminal nerves pathways. As evident from the dopamine and histopathology study of mice brain, nanoparticulate formulations could convincingly counteract haloperidol-induced Parkinsonism. These studies definitely indicated that the drug delivery system described here could offer an effective non-invasive approach to facilitate the access of BRC to the brain and more effectively in employing its formulations in therapeutic applications.
Declaration of interest
The authors are grateful to Department of Science and Technology (DST), New Delhi for providing DST INSPIRE Fellowship to S Md as financial assistance. This research work is original and all authors report no conflict of interest.
Notes
Related Research Data
Bibliography
- Luthra PM, Barodia SK, Raghubir R. Antagonism of haloperidol-induced swim impairment in l-dopa and caffeine treated mice: a pre-clinical model to study Parkinson's disease. J Neurosci Methods 2009;178:284-90
- Verma R, Nehru B. Effect of centrophenoxine against rotenone-induced oxidative stress in an animal model of Parkinson's disease. Neurochem Int 2009;55:369-75
- Muthane UB, Ragothaman M, Gururaj G. Epidemiology of Parkinson's disease and movement disorders in India: problems and possibilities. J Assoc Physicians India 2007;55:719-24
- Md S, Haque S, Sahni JK, et al. New non-oral drug delivery systems for Parkinson's disease treatment. Expert Opin Drug Deliv 2011;8(3):359-74
- Gárdián G, Vécsei L. Medical treatment of Parkinson's disease: today and the future. Int J Clin Pharmacol Ther 2010;48:633-42
- Dhuria SV, Hanson LR, Frey WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci 2010;99(4):1654-73
- Fazil M, Shadab M, Haque S, et al. Development and evaluation of rivastigmine loaded chitosan nanoparticles for brain targeting. Eur J Pharm Sci 2012;47(1):6-15
- Liu Z, Jiang M, Kang T, et al. Lactoferrin-modified PEG-co-PCL nanoparticles for enhanced brain delivery of NAP peptide following intranasal administration. Biomaterials 2013;34:3870-81
- Wen Z, Yan Z, Hu K, et al. Odorranalectin-conjugated nanoparticles: preparation, brain delivery and pharmacodynamic study on Parkinson's disease following intranasal administration. J Control Release 2011;151:131-8
- Vila A, Sanchez A, Tobio M, et al. Design of biodegradable particles for protein delivery. J Control Release 2002;78:15-24
- Haque S, Shadab M, Fazil M, et al. Venlafaxine loaded chitosan NPs for brain targeting, pharmacokinetic and pharmacodynamic evaluation. Carbohydr Polym 2012;89(1):72-9
- Ieva E, Trapani A, Cioffi N, et al. Analytical characterization of chitosan nanoparticles for peptide drug delivery applications. Anal Bioanal Chem 2009;393:207-15
- Luo Y, Zhang B, Whent M, et al. Preparation and characterization of zein/chitosan complex for encapsulation of tocopherol, and its in vitro controlled release study. Colloids Surf B Biointerfaces 2011;85:145-52
- Lim JH, Kim SS, Boo DH, et al. Protective effect of bromocriptine against BH4-induced Cath. A cell death involving up-regulation of antioxidant enzymes. Neurosci Lett 2009;451(3):185-9
- Md S, Khan RA, Mustafa G, et al. Bromocriptine loaded chitosan nanoparticles intended for direct nose to brain delivery: pharmacodynamic, pharmacokinetic and scintigraphy study in mice model. Eur J Pharm Sci 2013;48(3):393-05
- Nyholm D. Pharmacokinetic optimisation in the treatment of Parkinson's disease: an update. Clin Pharmacokinet 2006;45(2):109-36
- Degim IT, Acartürk F, Erdogan D, Lortlar ND. Transdermal administration of bromocriptine. Biol Pharm Bull 2003;26(4):501-5
- Choi YJ, Seo MK, Kim IC, Lee YH. High-performance liquid chromatographic assay of bromocriptine in plasma and eye tissue of the rabbit. J Chromatogr B Biomed Sci Appl 1997;694:415-20
- Valente D, Ezan E, Créminon C, et al. Enzyme immunoassays for bromocriptine and its metabolites. J Immunoassay 1996;17(4):297-20
- Salvador A, Dubreuil D, Denouel J, Millerioux L. Sensitive method for the quantitative determination of bromocriptine in human plasma by liquid chromatography–tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2005;820:237-42
- Faiyazuddin M, Ahmad N, Khar RK, et al. Stabilized terbutaline submicron drug aerosol for deep lungs deposition: drug assay, pulmonokinetics and biodistribution by UHPLC/ESI-q-TOF-MS method. Int J Pharm 2012;434:59-69
- Md S, Kumar M, Baboota S, et al. Preparation, characterization and evaluation of bromocriptine loaded chitosan nanoparticles for intranasal delivery. Sci Adv Mater 2012;4(9):949-60
- Dudhani AR, Kosaraju SL. Bioadhesive chitosan nanoparticles: preparation and characterization. Carbohydr Polym 2010;81:243-51
- Wilson B, Samanta MK, Santhi K, et al. Chitosan nanoparticles as a new delivery system for the anti-Alzheimer drug tacrine. Nanomedicine 2010;6(1):144-52
- Khan S, Patil K, Bobade N, et al. Formulation of intranasal mucoadhesive temperature-mediated in situ gel containing ropinirole and evaluation of brain targeting efficiency in rats. J Drug Target 2010;18(3):223-34
- ICH harmonised tripartite guideline. Validation of analytical procedures: text and methodology Q2(R1). ICH harmonised tripartite guideline 2005 [Last assessed 6 January 2014]
- ICH harmonised tripartite guideline. Stability testing of New Drug Substances and Products Q1A(R2). ICH harmonised tripartite guideline 2003 [Last assessed 6 January 2014]
- Kumar M, Misra A, Babbar AK, et al. Intranasal nanoemulsion based brain targeting drug delivery system of risperidone. Int J Pharm 2008;358(1-2):285-91
- Wang X, Chi N, Tang X. Preparation of estradiol chitosan nanoparticles for improving nasal absorption and brain targeting. Eur J Pharm Biopharm 2008;70(3):735-40
- Fenli S, Wang F, Zhu R, Li H. Determination of 5-hydroxytryptamine norepinephrine, dopamine and their metabolites in rat brain tissue by LC–ESI–MS–MS. Chromatographia 2009;69:207-13
- Trapani A, De Giglio E, Cafagna D, et al. Characterization and evaluation of chitosan nanoparticles for dopamine brain delivery. Int J Pharm 2011;419:296-07
- Nageswari A, Reddy KV, Mukkanti K. Stability-indicating UPLC method for determination of Imatinib Mesylate and their degradation products in active pharmaceutical ingredient and pharmaceutical dosage forms. J Pharm Biomed Anal 2012;66:109-15
- Al-Ghananeem AM, Saeed H, Florence R, et al. Intranasal drug delivery of didanosine-loaded chitosan nanoparticles for brain targeting; an attractive route against infections caused by aids viruses. J Drug Target 2010;18(5):381-8
- Zhu Z, Bartol M, Shen K, Johnson SW. Excitatory effects of dopamine on subthalamic nucleus neurons: in vitro study of rats pretreated with 6-hydroxydopamine and levodopa. Brain Res 2002;945:31-40
- Betarbet R, Sherer TB, MacKenzie G, et al. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 2000;3:1301-6
- Morris GA, Castile J, Smith A, et al. The effect of prolonged storage at different temperatures on the particle size distribution of tripolyphosphate (TPP)–chitosan nanoparticles. Carbohydr Polym 2011;84:1430-4
- Tsai YM, Chien CF, Lin LC, Tsai TH. Curcumin and its nano-formulation: the kinetics of tissue distribution and blood–brain barrier penetration. Int J Pharm 2011;416:331-8