
Abstract
Lowering low-density lipoprotein-cholesterol (LDL-C) is an effective strategy to reduce cardiovascular risk. However, a significant residual risk remains in statin-treated patients. High-density lipoprotein cholesterol (HDL-C) is a strong, independent and inverse predictor of cardiovascular risk in many epidemiologic studies and has, therefore, emerged as a potential novel therapeutic target for addressing this substantial residual risk. Nevertheless, the failure of cholesteryl ester transfer protein (CETP) inhibitors and niacin in clinical trials has generated considerable speculation about the beneficial effects of HDL. Experimental studies have identified several HDL cardioprotective functions, including enhancement of macrophage reverse cholesterol transport and endothelial function and its antioxidant, anti-inflammatory and anti-thrombotic properties. Furthermore, HDL is highly heterogeneous and the atheroprotective functions of the different HDL subpopulations are not completely understood. Current available data indicate that increased HDL-C levels do not always correlate with enhanced HDL functions and, therefore, should not be considered a biomarker of HDL functionality. The clinical application of the novel HDL-based therapeutic approaches requires the development of validated and reproducible measures of key HDL functions.
1. Introduction
Statin therapy reduces low-density lipoprotein cholesterol (LDL-C) levels and cardiovascular risk. Nonetheless, many patients have a significant residual risk, and additional therapeutic agents have been developed to increase high-density lipoprotein cholesterol (HDL-C) levels. The current concept that HDL protects against cardiovascular risk is based on abundant preclinical and epidemiological data. First, in multiple epidemiologic studies, HDL-C is inversely associated to cardiovascular risk Citation[1]. Second, low HDL-C remains a predictive outcome of major adverse cardiovascular events in patients treated with a high dose of statins and optimized LDL-C levels Citation[2], and a recent meta-analysis of 20 large randomized and controlled trials has demonstrated that low levels of HDL-C remain significantly and independently associated with an increased risk of myocardial infarction despite statin treatment Citation[3]. Third, the infusion of the main HDL protein component apoA-I or the mutant apoA-I Milano complexed with phospholipids (CSL-111 and ETC-216) reduces the coronary atheroma volume in small clinical studies Citation[4]. Fourth, preclinical analyses in genetically engineered mice demonstrate that increasing apoA-I levels prevent or regress atherosclerosis Citation[5].
2. Recent events challenge the causal relationship between HDL-C and cardiovascular risk
The recent publication of several clinical trials has cast doubts on the HDL-C hypothesis. Thus, the large outcome Atherothrombosis Intervention in Metabolic Syndrome with low HDL/High Triglycerides: Impact on Global Health Outcome (AIM-HIGH) trial was stopped prematurely after a follow-up of 3 years due to lack of cardiovascular benefit from the addition of niacin to statin therapy despite increasing HDL-C levels Citation[6]. However, it should be mentioned that simvastatin doses were adjusted to achieve a target LDL-C and the placebo group received higher doses of simvastatin compared with the niacin group; the former group also showed a surprising increase in HDL-C (12%), which may have minimized the HDL-raising benefits of niacin. Furthermore, the addition of extended-release niacin, combined with laropiprant to reduce flushing, in the Heart Protection Study 2: Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) did not reduce cardiovascular events in high-risk patients despite lowering LDL-C and triglyceride and raising HDL-C Citation[7]. Although the details have not been published yet, it is important to note that the data presented seem to point toward a better response in patients with higher LDL-C Citation[7]. It should be taken into account that these recent intervention trials were tested on top of mostly stringent statin therapy and this may have minimized the benefits of niacin compared with the positive effects of niacin therapy on the cardiovascular morbidity and mortality in the pre-statin era Citation[7,8].
The most potent novel drugs for raising HDL-C are cholesteryl ester transfer protein (CETP) inhibitors Citation[9]. However, the results of two large clinical trials using CETP inhibitors have also been disappointing Citation[9]. The Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial with patients receiving atorvastatin and torcetrapib worsened the primary combined cardiovascular end point compared with placebo after 12 months despite increasing HDL-C by 72% and reducing LDL-C by 25%. Furthermore, torcetrapib did not affect atheroma burden in three imaging trials Citation[9]. The worrisome results of these trials have been attributed, at least in part, to the off-target effects of torcetrapib, including an increase in serum aldosterone, thereby resulting in increased systolic blood pressure. The more recent dal-OUTCOMES study using the CETP inhibitor dalcetrapib raised HDL levels less effectively than torcetrapib and did not cause harmful effects; however, the trial was terminated in May 2012 for reasons of futility Citation[9]. The hypothesis of CETP inhibitor antiatherogenic effects is still being tested in two large cardiovascular clinical end-point trials with anacetrapib and evacetrapib Citation[8].
On the other hand, a large Mendelian randomization study showed that a single-nucleotide polymorphism (SNPs) in the endothelial lipase (EL) gene (LIPG Asn396Ser) associated with an important increase in HDL-C did not affect the risk of myocardial infarction Citation[10]. Furthermore, a 1 SD increase in HDL-C due to genetic score of 14 common SNPs was not associated with the risk of myocardial infarction, whereas a positive association between the risk of myocardial infarction and LDL-C due to genetic score was found Citation[10].
In light of the recent data, it is difficult to defend that elevated HDL-C levels per se reduce cardiovascular risk. It should be noted that low HDL-C associates strongly with elevated triglycerides which could explain the increased cardiovascular disease risk associated with low HDL-C, thereby suggesting that HDL-C could simply be a non-causal onlooker Citation[11]. Thus, an important question is how the findings on the HDL atheroprotective properties found in preclinical and population studies can be reconciled with the failure of niacin and CETP inhibitors in clinical trials and Mendelian randomization analyses.
3. HDL cholesterol increase does not always translate in enhanced HDL antiatherogenic functions
Historically, the reverse cholesterol transport (RCT) has been considered the main HDL antiatherogenic function but HDL has also antioxidant, anti-inflammatory, anti-thrombotic and endothelial-cell maintenance properties. It should be noted that the studies in genetically engineered animals indicate that the changes in macrophage-specific RCT and antioxidant and anti-inflammatory HDL activities are better predictors of atherosclerosis susceptibility than HDL-C Citation[5]. A good example is the scavenger receptor class-BI-deficient mice, which accumulate large quantities of HDL-C, but they have an impaired macrophage-to-feces RCT together with increased atherosclerosis susceptibility Citation[5]. Notably, the cholesterol-poor nascent HDLs (also named preβ-HDL) are the best acceptors of macrophage cholesterol efflux, the first critical step in RCT, which is consistent with the atheroprotective findings of short clinical studies that infuse recombinant apoA-I Citation[4]. In contrast, HDL from both torcetrapib- and niacin-treated patients has a normal or moderate increased macrophage cholesterol efflux Citation[9,12]; and dalcetrapib seems not to affect endothelial function Citation[9]. Taken together, these results indicate that therapeutic interventions must address not only HDL-C, but also the functional quality of these HDL particles.
4. What are the key metrics for assessing HDL function?
To date, the major clinical challenge is to obtain robust and reproducible assays of HDL functionality and surrogate markers of cardioprotective efficacy that can be applied to translational and clinical studies. A macrophage-to-feces RCT assay has been extensively applied in mice, but at present, it is difficult to quantify this RCT pathway in humans Citation[5,13]. In this regard, the most promising approach is to quantify macrophage cholesterol efflux ex vivo. One group used the cholesterol acceptor activity of apoB-containing lipoproteins depleted serum to demonstrate an inverse association between the ATP-binding cassette transporter (ABCA1)-dependent cholesterol efflux capacity and coronary artery disease independent of the HDL-C in three large patient cohorts Citation[14]. These findings were also found in a case–control group of patients referred to an ambulatory cardioprevention clinic, but not in a group of stable patients who underwent coronary angiography Citation[15]. Unexpectedly, these analyses showed that higher cholesterol efflux to apoB-depleted serum was associated with increased prospective risk of a composite cardiovascular end point of incident myocardial infarction, stroke, or death during 3 years of follow-up in the angiographic cohort Citation[15]. It should be taken into account that the cholesterol efflux assay only quantifies one component of the first-step RCT pathway without addressing the efficiency of the rest of the RCT steps or the other HDL cardioprotective functions. Indexes of monocyte chemotaxis, endothelial inflammation, HDL antioxidant activity or nitric oxide production may provide alternative approaches to measure HDL function Citation[16], but standardizing these methods will require a great deal of effort and their potential of clinical practice application remains uncertain, as they usually require HDL isolation as a first step. Recent findings indicate that the concentration of HDL particles is a good predictor of cardiovascular risk Citation[16]. Two approaches have been used for quantifying HDL number and size, nuclear magnetic resonance spectroscopy and ion mobility; however, these two methods have not been fully validated. It is of note that HDL is a highly heterogeneous lipoprotein which consists of > 50 proteins and 1000 different lipids. However, few studies have investigated the relationship between the HDL proteome and lipidome and the cardiovascular risk Citation[16]. A proteomic study found significant changes in the HDL protein cargo of patients with coronary artery disease and, in addition, the niacin/statin therapy shifts the HDL proteome from patients toward a healthy HDL profile Citation[16]. These findings emphasize the importance of additional research in this area and demonstrate that the vascular effects of HDL are not necessarily predicted by HDL-C levels, since cholesterol is only one lipid component of this lipoprotein.
5. Emerging HDL-based therapeutic agents
Beyond apoA-I/phospholipids infusion, the CETP inhibitors and niacin clinical trials, various apoA-I mimetic peptides (D-4F and L-4F) have been developed to mimic or even enhance the full-length apoA-I cardioprotective functions ( and ) Citation[4]. However, the impact of route of peptide delivery on efficacy remains unclear, and the subcutaneous administration of the most well studied apoA-I mimetic, L4F, did not improve HDL anti-inflammatory functions in two different Phase I clinical trials Citation[17]. An alternative HDL-targeted approach is direct focus in augmenting apoA-I levels through RVX-208, an orally available therapeutic agent that raises apoA-I production. The first analyses of the ApoA-I Synthesis Stimulation and Intravascular Ultrasound for Coronary Atheroma Regression Evaluation (ASSURE I) trial showed that RVX-208 treatment failed to find measurable benefits on plaque regression for patients with coronary artery disease and low HDL-C. However, further analysis of the ASSURE data found significant improvements in coronary atheroma measurements and major adverse cardiac events in patients with a high levels of serum high-sensitivity C-reactive protein (http://www.resverlogix.com/media/press_releases.html#2013). Intravenous infusion of recombinant lecithin:cholesterol acyltransferase (LCAT) APC-501 has been tested in patients with low LCAT activity and also in coronary artery disease patients and increased HDL-C Citation[8]. A carbamate-derived EL inhibitor has been reported to be effective in mice, but human studies have not been performed Citation[4]. Some new pathways affecting HDL metabolism may constitute promising therapeutic targets for improving HDL cardioprotective functions ( and ). As both ABCA1 and G1 are targeted for degradation by the microRNA (miR)-33, anti-miR-33 therapy resulted in increased HDL-C and macrophage cholesterol efflux in non-human primates Citation[13]. Furthermore, a role of cholesterol efflux pathways in the control of hematopoietic stem cells (HSCs) proliferation and the production of monocytes and neutrophils has been recently identified Citation[16]. Mice lacking ABCA1 and G1 displayed a significant increase in HSC mobilization and extramedullary hematopoiesis, which was suppressed by human apoA-I expression and liver X receptor (LXR) activation Citation[16]. Several LXR agonists (T0901317 and GW3965) reduce atherosclerosis in mice but, unfortunately, LXR promotes liver lipogenesis, which limits further clinical development Citation[18]. Recent findings also support the concept that the intestine may be an important determinant of RCT () Citation[18]. Therefore, intestine-specific LXR activation enhanced macrophage-to-feces RCT, reduced aortic atheroma volume and avoided the lipogenic toxicity associated with liver LXR activation in mice. The intestinal-specific LXR agonist GW6340 promotes macrophage RCT. Moreover, the gut microbiota can potentially affect the initiation of RCT via upregulating ABCA1 and G1 expression in macrophages by inhibition of a novel miRNA-10b signaling cascade Citation[18].
Figure 1. Schematic diagram of relevant pathways for HDL-based therapeutic interventions. ApoA-I is synthesized by liver and small intestine, acquires phospholipids and become nascent partially lipidated preβ-HDL particles. Preβ-HDL particles acquire free cholesterol from macrophages via adenosine triphosphate-binding cassette transporter (ABC) A-1. Free cholesterol is converted into cholesteryl ester within the HDL particle by the action of lecithin:cholesterol acyltransferase (LCAT), thereby resulting in mature HDL. The scavenger receptor type BI (SR-BI) and ABCG1 facilitate the cholesterol efflux process from macrophages to mature HDL. Endothelial lipase (EL) hydrolyzes HDL triglycerides and phospholipids, thereby remodeling larger HDL particles into smaller ones. HDL-cholesteryl ester can be transferred to VLDL or LDL by cholesteryl ester transfer protein (CETP) and returned to the liver through low-density lipoprotein cholesterol receptor (LDLR) or other receptors. The liver also selectively takes up HDL-associated cholesterol via SR-BI. The liver ABCG5/G8 heterodimer is involved in cholesterol transport to bile. Niemann-Pick C1-like 1 (NPC1L1) is of crucial importance for absorbing macrophage-derived cholesterol in the small intestine. Cholesterol may also be excreted back to the lumen by the intestinal ABCG5/G8 heterodimer. ABCA1 and ABCG1 are highly expressed in hematopoietic stem cells (HSCs) and negatively regulate the production of proatherogenic monocytes and neutrophils. Encircled targets for HDL-based therapeutic agents are indicated.
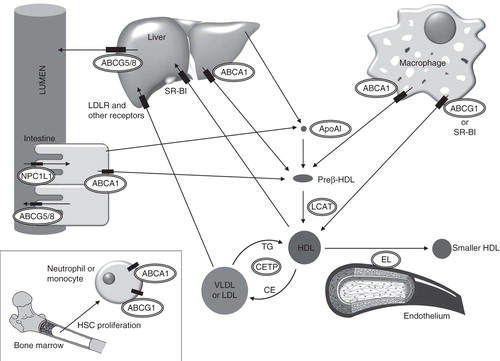
Table 1. HDL-based agents tested in humans or in preclinical models.
6. Conclusion
Although HDL-C levels are associated with decreased cardiovascular risk in epidemiological studies, the benefits of therapeutic HDL-C-raising are far from being confirmed. The current research is therefore focused on improving HDL function rather than simply targeting HDL-C levels. A major challenge is to develop clinical laboratory assays for quantifying HDL functions useful for predicting cardiovascular risk.
7. Expert opinion
Given the residual cardiovascular disease risk after targeting LDL-C levels, several novel therapeutic agents have been developed to either increase HDL-C or improve HDL atheroprotective functions. The recent clinical and genetic findings clearly indicate that interventions for raising HDL-C levels may not have positive effects in cardiovascular disease. As high HDL-C does not always correlate with enhanced HDL cardioprotective functions, HDL-C should not be considered a surrogate marker of HDL functionality.
The comprehensive assessment of HDL-raising drugs requires a precise quantification of HDL function. Furthermore, the recognition of the structural and functional heterogeneity of human HDL particles makes it difficult to establish which HDL subpopulations are responsible for the atheroprotective functions. Therefore, current research is moving toward the development of robust HDL function tests that can be widely applied in translational and clinical studies. In this context, it is also important to understand how HDL therapies affect HDL subpopulations, their lipidome and proteome cargo and their potential to lower cardiovascular risk. On the other hand, clinical outcome studies are necessary for evaluating some promising emerging HDL-based therapeutic agents, such as mimetic apoA-I peptides, intestinal LXR agonists and miR inhibitors. It should be noted that it took several decades to develop successful LDL-lowering therapies and these were also marked by controversy. Time will tell whether the novel HDL-targeted therapies are viable strategies for reducing cardiovascular disease.
Declaration of interest
This work was partly funded by European Cooperation in Science and Technology (COST) action BM0904, Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III, FIS 11-0176 (to F Blanco-Vaca) and FIS 12-00291 (to JC Escolá-Gil). CIBER de Diabetes y Enfermedades Metabólicas Asociadas is an Instituto de Salud Carlos III Project. The authors declare that there is no conflict of interest associated with this manuscript.
Acknowledgments
Although an important number of references were necessary for preparing this editorial and would be worthy of inclusion, we have mainly cited review manuscripts for making the manuscript feasible.
Bibliography
- Singh IM, Shishehbor MH, Ansell BJ. High-density lipoprotein as a therapeutic target: a systematic review. JAMA 2007;298:786-98
- Barter P, Gotto AM, LaRosa JC, et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med 2007;357:1301-10
- Jafri H, Alsheikh-Ali AA, Karas RH. Meta-analysis: statin therapy does not alter the association between low levels of high-density lipoprotein cholesterol and increased cardiovascular risk. Ann Intern Med 2010;153:800-8
- Degoma EM, Rader DJ. Novel HDL-directed pharmacotherapeutic strategies. Nat Rev Cardiol 2011;8:266-77
- Escola-Gil JC, Rotllan N, Julve J, Blanco-Vaca F. In vivo macrophage-specific RCT and antioxidant and antiinflammatory HDL activity measurements: new tools for predicting HDL atheroprotection. Atherosclerosis 2009;206:321-7
- Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011;365:2255-67
- Ginsberg HN, Reyes-Soffer G. Niacin: a long history, but a questionable future. Curr Opin Lipidol 2013;24:475-9
- Balder JW, Staels B, Kuivenhoven JA. Pharmacological interventions in human HDL metabolism. Curr Opin Lipidol 2013;24:500-9
- Barter PJ, Rye KA. Cholesteryl ester transfer protein inhibition as a strategy to reduce cardiovascular risk. J Lipid Res 2012;53:1755-66
- Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012;380:572-80
- Nordestgaard BG, Tybjaerg-Hansen A. Genetic determinants of LDL, lipoprotein(a), triglyceride-rich lipoproteins and HDL: concordance and discordance with cardiovascular disease risk. Curr Opin Lipidol 2011;22:113-22
- Khera AV, Patel PJ, Reilly MP, Rader DJ. The addition of niacin to statin therapy improves high-density lipoprotein cholesterol levels but not metrics of functionality. J Am Coll Cardiol 2013;62:1909-10
- Fisher EA, Feig JE, Hewing B, et al. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol 2012;32:2813-20
- Khera AV, Cuchel M, de la Llera-Moya M, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 2011;364:127-35
- Li XM, Tang WH, Mosior MK, et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol 2013;33:1696-705
- Rosenson RS, Brewer HB Jr, Ansell B, et al. Translation of high-density lipoprotein function into clinical practice: current prospects and future challenges. Circulation 2013;128:1256-67
- Watson CE, Weissbach N, Kjems L, et al. Treatment of patients with cardiovascular disease with L-4F, an apo-A1 mimetic, did not improve select biomarkers of HDL function. J Lipid Res 2011;52:361-73
- Lee-Rueckert M, Blanco-Vaca F, Kovanen PT, Escola-Gil JC. The role of the gut in reverse cholesterol transport–focus on the enterocyte. Prog Lipid Res 2013;52:317-28