
Abstract
Introduction: The application of computational tools to drug discovery helps researchers to design and evaluate new drugs swiftly with a reduce economic resources. To discover new potential drugs, computational chemistry incorporates automatization for obtaining biological data such as adsorption, distribution, metabolism, excretion and toxicity (ADMET), as well as drug mechanisms of action.
Areas covered: This editorial looks at examples of these computational tools, including docking, molecular dynamics simulation, virtual screening, quantum chemistry, quantitative structural activity relationship, principal component analysis and drug screening workflow systems. The authors then provide their perspectives on the importance of these techniques for drug discovery.
Expert opinion: Computational tools help researchers to design and discover new drugs for the treatment of several human diseases without side effects, thus allowing for the evaluation of millions of compounds with a reduced cost in both time and economic resources. The problem is that operating each program is difficult; one is required to use several programs and understand each of the properties being tested. In the future, it is possible that a single computer and software program will be capable of evaluating the complete properties (mechanisms of action and ADMET properties) of ligands. It is also possible that after submitting one target, this computer–software will be capable of suggesting potential compounds along with ways to synthesize them, and presenting biological models for testing.
1. Introduction
Currently, it is important to consider using informatics tools for drug design and virtual screening to obtain pharmacological products in short time frames Citation[1]. Computational resources reduce the time required to obtain drugs and decreases their associated economic costs, making it possible to propose potential drugs with low expenditure and selective targeting Citation[2]. Docking strategies have been used for several years to reveal drug mechanisms Citation[3]. Recently, docking strategies have been applied to drug prediction, which has resulted in the testing millions of compounds (virtual screening) to identify the best potential compounds for a specific target (protein) or to design multi-target compounds Citation[4]. However, to obtain data quickly, docking considers the target to be a rigid system, which may not yield reliable data Citation[5]. Therefore, several work groups have combined docking and molecular dynamics (MD) simulations by docking various protein conformations (snapshots) retrieved from MD simulations Citation[6] along with several protein conformations retrieved from the Protein Data Bank (PDB) Citation[7] for proteins that have been crystallized and solved several times Citation[8]. Using multiple protein structure conformations can provide more information about a drug’s binding and recognition properties. However, the docking procedure still lacks several biological properties such as the Gibbs free energy (ΔG) value that takes entropy properties into account; thus, several enhancements have been implemented to obtain more reliable data. Examples of this include improving the graphics processing unit hardware and algorithms that increase the number of simulations (scoring sampling), the calculation of ΔG (scoring functions), and the flexibility of the ligand and protein target Citation[9].
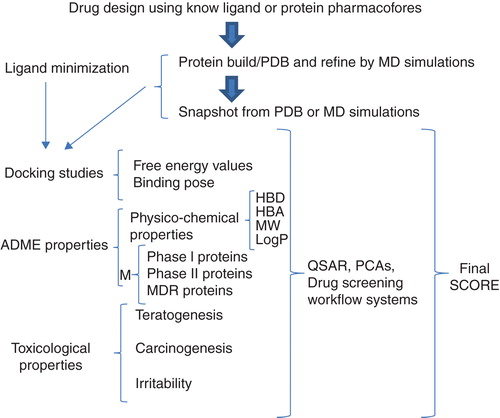
After the best compound is selected by combining protein structural studies (docking and MD simulation), the toxicity properties must be experimentally (in vitro and in vivo) evaluated () and the physicochemical characteristics of the compound must be determined because these properties affect the movement of compounds through biological liquids or cell membranes, which affects their efficacy and the duration of the compound’s administration. The physicochemical characteristics of compounds can be calculated by in silico procedures that have the advantage of saving time, and do not require animal models for adsorption, distribution, metabolism, excretion and toxicity (ADMET) determination. The physicochemical properties of compounds can be analyzed by applying Lipinski’s rule; however, there are tools available that can calculate ADMET parameters, to take into account all of the biological factors associated with biological actions on the drugs Citation[10]. One example of this type of tool is multiparameter optimization (MPO). In MPO, targets are located inside brain cells, where there are several biological barriers Citation[11]. In addition, there are new algorithms (for virtual screening workflow systems) that can be applied toward accelerating the automated search for potential compounds. It is suggested that drug researchers follow and consider all of the possible pharmacological properties of compounds () to ensure that their designed compounds reach their targets via oral administration without side effects. Metabolic properties are important to consider because they can inactivate a compound or yield toxic products. Additionally, to accelerate the search for potential compounds, there are many computational programs that help to calculate and predict ADMET properties and calculate the binding properties of ligands on targets. These data can then be robustly analyzed with quantitative structural activity relationship (QSAR), principal component analysis (PCA) and virtual screening workflow systems to obtain the leading compounds.
Figure 1. Theoretical strategy for obtaining the best drugs, including the target and the other pharmacological properties, through in silico studies.
2. Stages of drug discovery
2.1 First strategies for drug discovery
In the past, many drugs used to treat diseases were discovered by experiments conducted in animals or humans. Several compounds were obtained from natural sources, some of which have been used in diverse communities without any formal chemical characterization. However, due to technological advances, chemical isolation and characterization have identified the compounds responsible for biological reactions. Using this knowledge, it has been possible to obtain improved compounds by making specific chemical modifications to improve their biological properties, such as pharmacodynamics and pharmacokinetics, or to decrease their toxicity Citation[2]. The first strategies involved chemical syntheses and biological assays that resulted in potential drugs for use in human diseases. Currently, computational resources have made it possible to reproduce many biological success stories by applying a vast number of ADMET descriptors as well as providing insights leading to new potential compounds using computational algorithms Citation[10,12] that can then be experimentally validated. In this review, we focus on depicting the advantages of using automatized computational programs capable of evaluating several million compounds (either designed or retrieved from known data bases) considering the entirety of biological factors involved in their pharmacological actions and ADMET properties that eventually inform their mechanisms of action at their biological targets and pharmacokinetic properties, respectively. We will focus on explaining the initial applications of computational resources up to the present, where computational resources have evidently grown as a result of new technological advances and computational algorithms.
2.2 First applications of computational tools to drug discovery
Initially, computational tools were employed for studying DNA or protein sequences using text analyzers such as Perl programs. Next, several friendly online resources appeared that were important for studying evolutionary behavior, mutation, drug resistance and other biological properties. In addition, after generating three-dimensional (3D) DNA structures, several proteins were characterized and deposited at PDB, which could be studied structurally or used to build homologous proteins for drug design or virtual screening Citation[13]. In addition, computational tools have helped visualize molecules in 2 dimensions (2D), as well as in 3D, for evaluating the molecule structure space with molecular mechanics or force fields. Another important field that used these computational tools is molecular visualization. The aforementioned computational resources are still in use and additional resources are on the rise, compiling more advanced theoretical data on the protein motion (MD simulations) and electronic factors (e.g., HOMO: the highest occupied molecular orbital, and LUMO: the lowest unoccupied molecular orbital) of proteins and small ligands. By coupling the above-mentioned strategies together with ADMET evaluation Citation[14], one can obtain complete data for drug design (), with the great possibility of finding potential compounds that can pass their preclinical and clinical phases and be made available to human patients.
2.3 Docking methods for drug discovery
Three-dimensional models of proteins obtained from the PDB or constructed using homology modeling or ab initio prediction are used to describe the mechanisms of action of drugs and for drug design. By using 3D models, one can evaluate different compounds with docking methods that describe the molecular surface properties of small molecules and/or protein targets. Initially, docking was performed manually, and small molecules were coupled to rigid proteins to calculate their nonbonding interactions and the ΔG values of the complex exclusive of entropy properties. There are several programs available for docking studies, such as DOCK Citation[15], FlexX Citation[16], GLIDE Citation[17], ICM Citation[18], PhDOCK Citation[19], Surflex Citation[20], Autodock Citation[21] and GOLD Citation[22], among others, and additional programs have been designed for virtual screening by obtaining binding and ΔG values efficiently Citation[15], for example, AutoDock-Vina Citation[23], DockoMatic Citation[24], PyRx Citation[25], DockingServer Citation[26] and MOLA Citation[27], among others. Although there are several programs Citation[28] available, each of them with their loyal adherents, one of the most popular programs is Autodock because it is free. Although docking is a widely employed and useful computational tool, it has several limitations. For example, some docking studies allow for ligand flexibility while keeping the protein rigid. Despite the fact that docking studies help predict the potential ligands for protein targets, it is possible that this predicted behavior is not identical to that found in nature. This is because docking methods do not take several natural factors into account such as water molecules and protein motion. Consequently, some researchers are focused on incorporating specific types of protein motion into docking such as the motion of side chain residues or induced fit binding. However, these strategies neglect the distant structural protein components that could affect ligand recognition Citation[29]. Thus, researchers have focused on using docking and MD simulations for obtaining the most accurate free energy values and binding poses Citation[8].
2.4 MD simulations for drug discovery
MD simulations help to account for the biological behavior of proteins in aqueous environments or lipid membranes and allow the proteins to undergo natural motion, adopting various structural conformations that can be used for docking studies Citation[30]. However, it is very difficult to find identical binding free energy values and binding poses with several structural target conformers using MD simulations. Still, with these strategies, one can obtain average values and poses, thus yielding more reliable data than those obtained solely by docking. Despite the time-consuming nature of MD simulations Citation[8], it can yield more reliable data on potential compounds with the ADMET properties that comply with drug requirements (). MD simulations data can be employed to retrieve several snapshots (of natural behavior) that can be used to test ligands by docking or virtual screening Citation[6]. There are several programs for MD simulations such as GROMACS Citation[31], AMBER Citation[32] and NAMD Citation[33]. NAMD has been one of the most popular programs because it is free for academic use and is user-friendly.
2.5 ADMET properties
It is important to mention that although a combination of docking and MD simulations will yield reliable data when searching for the best ligand for a target, other factors outside the binding site, such as ADMET properties, are important for a drug to reach its target. Due to automated computational tools, it is now possible to obtain ADMET properties using in silico methods Citation[34], which not only selects the ligand with the best affinity for a target but also predicts toxicity properties and absorption parameters as well as potential metabolites from the biotransformation phase Citation[34], avoiding those compounds that undergo significant structural changes or are susceptible to the multiple drug resistance (MDR) system (). Therefore, several research groups are taking ADMET properties into consideration for obtaining drugs that could be successful if their pharmacodynamic and pharmacokinetic properties were maintained within the allowed limits for the ADMET properties using molecular modeling to suggest the conceivable mechanisms Citation[2]. A possible method () for selecting the most promising ligands is as follows: the ligands are designed or retrieved from data bases, and then the ligands are submitted to MOLINSPIRATION Citation[35] and OSIRIS property explorer Citation[36] to evaluate their physicochemical and toxicity properties, and finally, the ligands are evaluated for additional properties that are similar to ADMET properties, for which VolSurf Citation[34] is recommended, and the ligand’s metabolism is predicted using MetaSite Citation[34]. Due to important improvements to computational hardware and software, it is now possible to evaluate molecular motion. In addition, quantum chemistry has made it possible to evaluate the electronic properties of molecules to obtain several hundreds of molecular descriptors for QSAR Citation[30] or PCA studies to calculate ADMET properties, which can be used for enriching the scores for selecting the lead compounds Citation[14].
3. Conclusion
In conclusion, it is important to mention that all of the computational methods that are available to evaluate ligand–protein interaction and ADMET properties have had an important impact on drug design. Because these tools enable the timely evaluation of millions of compounds for obtaining potential drugs to treat several human diseases with low costs in terms of human and economic resources, patients will pay less for these drugs than for those obtained by traditional procedures, such as synthesis and biological assays, that do not address the properties of potential drugs, such as their interactions on biological targets, biological barrier crossing, metabolism avoidance and nontoxicity.
4. Expert opinion
Computational resources contain structural information on ligands and biological targets, which helps drug designers to develop new compounds using rational strategies for obtaining potentially useful ligands. In addition, drug design with rational strategies allows for the discovery of compounds that are capable of crossing biological barriers without toxicity Citation[1,2]. In diseases with different targets, it is possible to use computational tools to design multi-target compounds to avoid drug resistance, achieve multiple therapeutic uses and increase pharmacological efficacy. After new compounds are designed in view of the pharmacophore information from the ligand or target, the principal functional groups must be taken into consideration. This process can be facilitated by several programs Citation[28] using efficient automated servers Citation[37]. Next, one can achieve virtual screening by applying docking studies using Autodock Vina Citation[33] or one of the other previously mentioned programs (). Consequently, MD simulations could improve the quality of the data obtained because they take the structural motion of the targets and ligands into account Citation[9]. Despite the fact that docking and MD simulations are very important for the in silico evaluation of new potential drugs, these methodologies are based on force fields, thus, they do not allow one to obtain the electronic properties of newly designed drugs. Using force fields alone allows one to obtain results in a short time period by testing millions of compounds; however, using electronic parameters to obtain structural details allows one to design compounds with more rational strategies. For example, quantum mechanics (QM) can be employed to combine docking, MD simulations and QM Citation[30]. If the structural information with respect to atomic and electronic behavior is obtained, it is possible to obtain the structural binding poses and movements as well as the electronic details that help describe the recognition process under negative (HOMO) and positive charges (LUMO), exposing those interactions that can be difficult to identify with nonbonded interactions. These theoretical tools are time-consuming; however, one can apply QSAR and PCA studies for analyzing ADMET, binding (ΔG) and biological properties, if they exist Citation[14], which yield information from the ligands and target more efficiently than molecular modeling Citation[30]. The problem is that QSAR and PCA analyses do not depict the structural details involved in ligand recognition. However, by combining docking, MD simulations, QM, QSAR and PCA studies, one could obtain significant results during the design and evaluation of a new potential drug Citation[30]. Furthermore, one can begin with QSAR studies and PCAs using biological information retrieved from web servers or by molecular modeling, including docking, MD simulations and QM data, and later include physicochemical ADMET-related properties (information that can be obtained from virtual servers) because one can obtain molecular information via Lipinski’s rule as well as other physicochemical properties that are more accurate with respect to clinical data Citation[11]. This allows one to infer which new compounds would be capable of crossing biological barriers and would not be metabolized or accessed from the MDR. In addition, one can measure toxicity properties and discern whether a new compound could produce carcinogenesis, teratogenesis and/or irritability Citation[35]. Finally, with the parameters obtained by physicochemical and toxicological evaluation as well as the metabolism patterns predicted and studied with molecular modeling (docking, MD simulations), it is possible to assign a score to select the best potential compound (). However, it is important to mention that there are recent computational tools (screening workflow systems) that expand the search for potential compounds using advanced algorithms such as Taverna Citation[38] KNINE Citation[39] and Pipeline Pilot Citation[40], which allow researchers to obtain potential compounds in an efficient manner. These resources can be applied to designed molecules or coupled to the databases of small ligands such as PubChem Citation[41], ChemSpider Citation[42], CheMBL Citation[43] and the published data at OpenTox Citation[44], Chembench Citation[45] and OCHEM Citation[46], among others, to explore several biological properties in order to test ligands that are reported elsewhere. Due to the success of these theoretical algorithms, which are capable of yielding reproducible data, several programs have been developed that have increased the number of publications in specialized journals Citation[1]. One problem is that many programs are handled independently and numerous physical and chemical descriptors used for QSAR are poorly understood for 3D models. For that reason, it will be necessary in the future to combine molecular modeling with QSAR, and then perform further analyses with PCA and screening workflow systems to inform chemists regarding the most promising compounds. It is possible that there will be one program in a single computer that explores the complete biological properties of compounds, including their biological action prior to ADMET properties. Alternatively, programs could include algorithms that, upon submitting a target, would be capable of yielding potential compounds with improved ADMET properties and include ways for them to be synthesized as well as suggest the biological experiments needed to corroborate the biological properties. Subsequently, one could focus on synthesizing and testing the compounds for validation purposes using experimental assays. Thus, the in vitro and in vivo evaluations of a new compound obtained from in silico evaluations allow one to conserve economical and temporal resources for obtaining potential drugs. In the future, these strategies could be applied toward several human diseases to great advantage, yielding lower prices due to the fewer resources needed to identify the drugs, and with the distinct possibility of discovering selective or multi-target drugs with negligible side effects.
Acknowledgements
The authors have had their language checked through the American Journal Experts (AJE).
Declaration of interest
The authors are supported by the Instituto Politécnico Nacional, the Programa Institucional de Formacion de Investigadores (PIFI) and the Secretaria de Investacion y Posgrado (SIP). MC Rosales-Hernandez is supported by the Comision de Operacion y Fomento de Actividades Academias (COFAA), the Estimulo al Desempeno a la Investigacion (EDI) and MEDIX (Empresa Mexicana de se Medicamentos). J Correa-Basurto is also supported by the Consejo Nacional de Ciencia y Technologia grant no. 132353 (CONACYT) and the Programa Iberoamericano de Ciencia y Technologia para el Desarrollo No 214RT0842 (CYTED). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Notes
Bibliography
- Tetko IV. The perspectives of computational chemistry modeling. J Comput Aided Mol Des 2012;26:135-6
- Cumming JG, Davis AM, Muresan S, et al. Chemical predictive modelling to improve compound quality. Nat Rev Drug Discov 2013;2:948-62
- Levoin N, Blondeau C, Guillaume C, et al. Elucidation of the mechanism of inhibition of cyclooxygenases by acyl-coenzyme A and acylglucuronic conjugates of ketoprofen. Biochem Pharmacol 2004;68:1957-69
- Plewczynski D, Łaźniewski M, Augustyniak R, et al. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J Comput Chem 2011;32:742-55
- Baxter CA, Murray CW, Waszkowycz B, et al. New approach to molecular docking and its application to virtual screening of chemical databases. J Chem Inf Comput Sci 2000;40:254-62
- Bermúdez-Lugo JA, Perez-Gonzalez O, Rosales-Hernández MC, et al. Exploration of the valproic acid binding site on histone deacetylase 8 using docking and molecular dynamic simulations. J Mol Model 2012;18:2301-10
- PDB. Available from: http://www.rcsb.org/pdb/home/home.do
- Ramírez-Durán LA, Rosales-Hernández MC, Hernández-Rodríguez M, et al. Mapping myeloperoxidase to identify its promiscuity properties using docking and molecular dynamics simulations. Curr Pharm Des 2013;19:2204-15
- Sinko W, Lindert S, McCammon JA. Accounting for receptor flexibility and enhanced sampling methods in computer-aided drug design. Chem Biol Drug Des 2013;81:41-9
- Cheng F, Li W, Liu G, et al. In silico ADMET prediction: recent advances, current challenges and future trends. Curr Top Med Chem 2013;13:1273-89
- Wager TT, Hou X, Verhoest PR, et al. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci 2010;1:435-49
- Shultz MD. Setting expectations in molecular optimizations: strengths and limitations of commonly used compositeparameters. Bioorg Med Chem Lett 2013;23:5980-91
- Petrey D, Xiang Z, Tang CL, et al. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homologymodeling. Proteins 2003;53:430-5
- Faller B, Ottaviani G, Ertl P, et al. Evolution of the physicochemical properties of marketed drugs: can history foretell the future? Drug Discov Today 2011;16(21-22):976-84
- DOCK blaster. Available from: http://blaster.docking.org/ [Last accessed 18 December 2014]
- BioSolveIT GmbH FlexX. Available from: http://www.biosolveit.de/flexx/ [Last accessed 18 December 2014]
- Schrodinger’s Glide. Available from: http://www.schrodinger.com/Glide/ [Last accessed 18 December 2014]
- Molsoft. ICM docking and screening. Available from: http://www.molsoft.com/docking.html [Last accessed 18 December 2014]
- iScreen. Available from: http://iscreen.cmu.edu.tw/phdock.php [Last accessed 18 December 2014]
- Tripos. Surflex-Dock. Available from: http://www.tripos.com/index.php?family=modules,SimplePage,,,&page=surflex_dock&s=0 [Last accessed 18 December 2014]
- AutoDock. Available from: http://autodock.scripps.edu/ [Last accessed 18 December 2014]
- Gold Suite. Available from: https://www.ccdc.cam.ac.uk/Solutions/GoldSuite/Pages/GOLD.aspx [Last accessed 18 December 2014]
- AutoDock Vina. Available from: http://vina.scripps.edu/ [Last accessed 18 December 2014]
- DockoMatic. Available from: http://dockomatic.soft112.com/ [Last accessed 18 December 2014]
- Python prescription virtual screening tool. Available from: http://pyrx.sourceforge.net/ [Last accessed 18 December 2014]
- DockingServer. Available from: http://www.dockingserver.com/web [Last Accessed 18th December 2014]
- BioChemCore MOLA. Available from: http://www.esa.ipb.pt/biochemcore/index.php/ds/m [Last accessed 18 December 2014]
- Swiss Institute of Bioinformatics. Click2Drug. Available from: http://www.click2drug.org/directory_StructureBasedScreening.html [Last Accessed 18th December 2014]
- Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins 2006;65:15-26
- Correa-Basurto J, Bello M, Rosales-Hernández MC, et al. QSAR, docking, dynamic simulation and quantum mechanics studies to explore the recognition properties of cholinesterase binding sites. Chem Biol Interact 2014;209:1-13
- Gromacs. Available from: http://www.gromacs.org/ Last accessed 18th December 2014]
- Amber. Available from: http://ambermd.org/ Last accessed 18th December 2014]
- NAMD. Available from: http://www.ks.uiuc.edu/Research/namd/ [Last accessed 18 December 2014]
- VolSurf+. Available from: http://www.moldiscovery.com/software/vsplus [Last accessed 18 December 2014]
- Molinspiration Cheminformatics Software. Available from: http://www.molinspiration.com/ [Last accessed 18 December 2014]
- OSIRIS property explorer. Available from: http://www.organic-chemistry.org/prog/peo/ [Last accessed 18 December 2014]
- Computational Tools for Drug Discovery. Available from: http://crdd.osdd.net/admet.php [Last Accessed 18th December 2014]
- Taverna. Available from: http://www.taverna.org.uk/ [Last accessed 18 December 2014]
- KNIME server. Available from: http://www.knime.org/knime-server [Last accessed 18 December 2014]
- BIOVIA pipeline pilot overview. Available from: http://accelrys.com/products/pipeline-pilot/ [Last accessed 18 December 2014]
- PubChem. Available from: https://pubchem.ncbi.nlm.nih.gov/ [Last accessed 18 December 2014]
- ChemSpider. Available from: http://www.chemspider.com/ [Last accessed 18 December 2014]
- ChEMBL. Available from: https://www.ebi.ac.uk/chembl/ [Last accessed 18 December 2014]
- OpenTox. Available from: http://www.opentox.org/ [Last accessed 18 December 2014]
- Chembench. Available from: https://chembench.mml.unc.edu/ [Last accessed 18 December 2014]
- OCHEM. Available from: https://ochem.eu/home/show.do