Abstract
Huntington's disease (HD) is a devastating autosomal-dominant neurodegenerative condition caused by a CAG repeat expansion in the gene encoding huntingtin which is characterised by progressive motor impairment, cognitive decline and neuropsychiatric disturbances. There are currently no disease-modifying treatments available to patients, but a number of therapeutic strategies are currently being investigated, chief among them are nucleotide-based ‘gene silencing' approaches, modulation of huntingtin post-translation modification and enhancing clearance of the mutant protein. In 2008, the authors' review highlighted the need to develop and validate biomarkers and provided a systematic head-to-head comparison of such measures. They searched the PubMed database for publications, which covered each of the subheadings mentioned below. They identified from these list studies which had relevance to biomarker development, as defined in their previous review. Building on a tradition of collaborative research in HD, great advances have been made in the field since that time and a range of outcome measures are now being recommended in order to assess efficacy in future therapeutic trials.
1. Biomarkers for clinical trials
1.1 Neuroimaging
It has long been established that neuroimaging can demonstrate changes within the brain in both early Huntington's disease (HD) subjects and premanifest gene carriers, with both regional and global measures sensitive to change from the very early stages. There is now increasing recognition of the role of white matter in the degenerative process, with widespread change detectable more than 15 years before expected disease onset Citation[1,2]. Thanks to large-scale studies such as TRACK-HD Citation[1] and PREDICT-HD Citation[2], there is now evidence that these imaging metrics are able to show reproducible group differences across multiple sites and that change over time can be demonstrated robustly in this slowly progressive disease over periods as short as 12 months, a timeframe which is realistic for clinical trials Citation[3]. The greatest effect sizes have been reported using rates of caudate loss, with global measures also demonstrating great power to detect disease-related change, providing sample sizes which could feasibly be recruited for therapeutic trials Citation[4]. TRACK-HD has also demonstrated the primacy of cross-site standardisation and robust quality control measures to ensure consistency of these data.
More recently, imaging modalities other than structural magnetic resonance imaging (MRI) have been proposed as suitable candidates for clinical trials. Diffusion weighted imaging (DWI), which specifically investigates the structural integrity of white matter, has demonstrated change over time in early HD subjects Citation[5], although this finding was not reproduced by a larger study Citation[6]. Functional MRI (fMRI) has the potential to detect disease-related variation in brain activity, showing cross-sectional differences prior to disease onset Citation[7]. However, a recent study failed to show change over a 2-year period Citation[8]. While fMRI and DWI remain highly promising candidates for probing dysfunctions in neural networks that are the hallmark of premanifest HD, further work is required to investigate their sensitivity to longitudinal change as well as validate measures across multiple sites before the clinical utility of these modalities are established.
Other recent work has shown cross-sectional disease-related differences in motor cortex excitability using transcranial magnetic stimulation Citation[9]. Altered metabolite profiles, suggestive of neuronal dysfunction have also been reported in early HD and treatments aiming to improve neuronal health might effectively be assessed using such a metric Citation[10]. Again, further cross-site validation and follow-up data are required to assess the utility of these modalities.
1.2 Quantitative clinical measures
Clinical ratings scales remain the most widely used measures of progression in HD. However, due to their large inter-rater variability and floor and ceiling effects, recent work has focused on more objective techniques which may better capture the range of deficits in both early and premanifest stages of disease.
Quantitative motor assessments such as finger tapping, grip force and tongue protrusion tasks have shown significant cross-sectional effects across the disease spectrum Citation[1]. These measures have also demonstrated robust change over time in early HD subjects Citation[3,11]; effect sizes suggest such metrics may prove useful as outcome measures in clinical trials Citation[4]. They are relatively insensitive over short time intervals in the premanifest stage showing small effect sizes longitudinally Citation[11], however, over a 36-month interval these measures look more promising as outcome measures for trials in premanifest subjects (TRACK-HD, unpublished data). Similarly, oculomotor and neuropsychiatric assessments which show cross-sectional disease-related deficits Citation[1], appear to be relatively insensitive to longitudinal change over periods of up to 24 months Citation[4]. However, some neuropsychiatric measures show progression over 36 months (TRACK-HD, unpublished data).
Numerous studies have shown cross-sectional cognitive deficits, even many years before disease onset Citation[1,2]. Longitudinal studies have shown change over time in early HD, although effect sizes vary considerably depending on the specific cognitive test employed Citation[4,12]. Sensitivity to change in the premanifest stages is again very limited over 24 months Citation[4], but after 36 months changes in cognition become apparent in this cohort (TRACK-HD, unpublished data).
1.3 Biofluids
Robust, pathologically relevant changes in accessible biofluids such as blood remain highly desirable. The pathways previously highlighted as suggesting potential biomarkers remain of interest, particularly oxidative, metabolic and inflammatory markers. These require testing in samples derived from multiple populations and over time, to establish their potential utility for future clinical trials. Newly identified biomarker candidates that require further investigation include cholesterol metabolites Citation[13] and indirect markers of transcriptional dysregulation such as the histone component H2AFY Citation[14]. Meanwhile clusterin, which the authors' work revealed as a possible HD plasma biomarker Citation[15], has since been highlighted as a major genetic modifier and possible blood biomarker of Alzheimer's disease Citation[16].
Accurately measuring levels of mutant (mHtt) and wild-type huntingtin in biosamples will be essential for testing therapeutics aimed at reducing mHtt levels, including promising nucleotide-based ‘gene silencing' treatments expected to enter clinical trials in the near future, as well as drugs aimed at altering the handling and removal of mHtt through post-translational modification and proteolysis. Existing Htt assays have suffered from low sensitivity and have been restricted to aggregated mHtt. A recently described agarose gel electrophoresis assay (AGERA) has lowered the threshold for aggregate detection and quantification Citation[17], but the role of aggregates is debated and it is likely that soluble oligomeric mHtt is most toxic. Until recently, the detection of soluble Htt was limited by the intrinsic insensitivity of assays, and a lack of suitable antibodies. Novel antibodies and a fluorescence-enhanced antibody assay using time-resolved Förster resonance energy transfer (TR-FRET) has now yielded a reliable, highly sensitive method of quantifying both mutant and total huntingtin in blood samples Citation[18]. This assay is likely to empower future clinical trials. Meanwhile, attempts are underway to develop assays that are even more sensitive and scalable.
Given its close biochemical and physical relationship with the brain, and utility in other neurodegenerative diseases as a source of biomarkers, cerebrospinal fluid (CSF) has natural appeal, despite being time-consuming and more invasive than phlebotomy. Efforts to identify CSF biomarkers for HD have been hampered by a shortage of samples and lack of consistency between existing small collections. In one major study, proteomic data generated from one sample set by five laboratories were integrated, revealing changes (mainly reductions) in many brain-specific proteins, but raising concerns about consistency between laboratories Citation[19]. Pursuing a hypothesis-driven approach, another group has identified neurofilament light chain and tau as potential neuron-specific CSF markers Citation[20]; but neither provides complete separation from controls or has been studied longitudinally. Multisite longitudinal CSF collection studies are in the planning stage.
2. Future directions
2.1 Neuroimaging
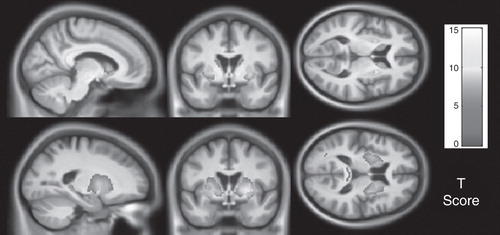
It is clear from studies such as TRACK-HD and PREDICT-HD that there are striking neuropathological changes up to 15 years prior to the onset of manifest disease Citation[1,2] (). Nevertheless, these subjects maintain their performance on functional measures. It would appear that there is a compensatory mechanism within the brain which finally breaks down at disease onset Citation[7]; imaging modalities other than structural MRI will be required to elucidate this process. Track-On HD seeks to apply novel imaging techniques across multiple sites, following the premanifest cohort from the TRACK-HD study. Whether these imaging modalities are able to produce metrics which are robust and sensitive enough to be used in clinical trials is yet to be determined.
Figure 1. Parametric maps showing regions with statistically significant atrophy over 24 months in grey (top) and white matter (bottom) in far-from-onset premanifest subjects compared with controls. Results were adjusted for age, sex, study site and scan interval and are corrected for multiple comparisons with familywise error at the p < 0.05 level. Figure shown courtesy of the TRACK-HD study.
A means of quantifying wild-type and mutant huntingtin in the brain would be a valuable tool for demonstrating central nervous system (CNS) target engagement of huntingtin-lowering therapies in the main organ of interest, especially since early gene silencing therapeutics are likely to be delivered directly into the CNS rather than systemically. Work is underway to develop and test PET (positron emission tomography) ligands that will enable visualisation of huntingtin load, as a predictive tool and a means of assessing response to therapy.
2.2 Quantitative clinical measures
Further refinement and validation is required if objective quantitative measures of motor and cognitive function are to demonstrate sensitivity to premanifest disease, or be accepted as outcome measures in clinical trials. It is likely that clinical trials in premanifest subjects will need to be at least 36 months in duration if current functional and clinical outcome measures were to be used. A number of cognitive tasks show promise as biomarkers, particularly measures of visuospatial integration Citation[3]. A cognitive ‘toolkit' for premanifest and manifest HD, which will build on the existing evidence, is nearing completion Citation[21]. Neuropsychiatric measures are likely to be included in future clinical trials since assessments of patient well-being are of paramount importance, but these too require the development of more reproducible and sensitive methodology. In addition to these initiatives, ongoing longitudinal studies such as PREDICT-HD and Track-On HD seek to optimize measures in these different functional modalities.
2.3 Biofluids
Novel assays to measure soluble mutant huntingtin, and their refinement for high-throughput use, are likely to emerge as one of the most important developments in the field and will become a standard measure for clinical trials Citation[18]. The use of CSF as the biofluid of choice for interrogating the CNS milieu is likely to occur in HD as it has in other neurodegenerative diseases, but standardised, multisite, longitudinal CSF natural history studies are needed Citation[22]. Finally, the most promising biomarker candidates need to be studied ‘head-to-head' using the large longitudinal blood sample collections that studies such as TRACK-HD have generated Citation[4].
Though a decade of research has not yielded robust biochemical biomarkers of HD, progress has been made. A consensus is emerging that the detection of neuron-specific proteins in blood may be an unrealistic aim, but since the mutant huntingtin protein is expressed ubiquitously, that does not mean that tracking change in peripheral tissues lacks merit. What is needed is scrupulous study of how any measure is involved in HD pathogenesis, and judicious interpretation of changes seen in the context of therapeutic trials.
3. Expert opinion
Considerable progress has been made towards identifying biomarkers which show promise as outcome measures in clinical trials. Multinational studies such as PREDICT-HD and TRACK-HD have proved invaluable in validating measures which are robust and reproducible across different sites and cultures. The most recent publication from the TRACK-HD study provides a direct comparison of the effect sizes for a range of modalities, and outlines a battery suitable for use in clinical trials in early HD Citation[4]; this includes structural neuroimaging, quantitative motor, cognitive and neuropsychiatric assessments. However, as yet such a battery does not exist for a premanifest cohort as current functional measures appear to be relatively insensitive at this disease stage over a 24-month time period. The 36-month dataset from TRACK-HD is currently being analysed and it is anticipated that this may improve the battery for clinical trials in the premanifest cohort. Neuroimaging demonstrates the largest effect sizes, but further investigation of the relationship between such metrics and functional performance is required. There is clearly a need for robust functional assessments to be included in any trial and there are now a number of candidate biomarkers from different modalities which can feasibly be implemented. The individual elements of this battery selected for a particular trial will of course depend on the proposed mode of action of therapy, timeframe and cohort. It was previously predicted that ultimate evaluation of biomarker candidates would need to take place within the framework of clinical trials of disease-modifying treatments. This model for biomarker development is now deployed in HD clinical trials (e.g., mHtt and ILs are being measured as part of the PADDINGTON study of sirtuin-1 inhibition) and, though challenging, this approach is essential, since it is only when disease, biomarker and drug are studied together that the true interplay between them can emerge.
Declaration of interest
The research in SJ Tabrizi's laboratory is funded by MRC, BBSRC, High Q/CHDI Foundation, EU FP7 Framework, UCL/UCLH NIHR Biomedical Research Centre, UK HD Association, European HD Network and the UK Dementia and Neurodegenerative Diseases Network. EJ Wild is supported by the National Institutes for Health Research and the Academy of Medical Sciences, UK. UCLH/UCL receives a proportion of its funding from the Department of Health's NIHR Biomedical Research Centres funding scheme.
Bibliography
- Tabrizi SJ, Langbehn DR, Leavitt BR, Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol 2009;8(9):791-801
- Paulsen JS, Langbehn DR, Stout JC, Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 2008;79(8):874-80
- Tabrizi SJ, Scahill RI, Durr A, Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol 2011;10(1):31-42
- Tabrizi SJ, Reilmann R, Roos RA, Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol 2012;11(1):42-53
- Weaver KE, Richards TL, Liang O, Longitudinal diffusion tensor imaging in Huntington's disease. Exp Neurol 2009;216(2):525-9
- Sritharan A, Egan GF, Johnston L, A longitudinal diffusion tensor imaging study in symptomatic Huntington's disease. J Neurol Neurosurg Psychiatry 2010;81(3):257-62
- Kloppel S, Draganski B, Siebner HR, Functional compensation of motor function in pre-symptomatic Huntington's disease. Brain 2009;132(Pt 6):1624-32
- Wolf RC, Sambataro F, Vasic N, Longitudinal functional magnetic resonance imaging of cognition in preclinical Huntington's disease. Exp Neurol 2011;231(2):214-22
- Schippling S, Schneider SA, Bhatia KP, Abnormal motor cortex excitability in preclinical and very early Huntington's disease. Biol Psychiatry 2009;65(11):959-65
- Weir DW, Sturrock A, Leavitt BR. Development of biomarkers for Huntington's disease. Lancet Neurol 2011;10(6):573-90
- Rowe KC, Paulsen JS, Langbehn DR, Self-paced timing detects and tracks change in prodromal Huntington disease. Neuropsychology 2010;24(4):435-42
- Beglinger LJ, Duff K, Allison J, Cognitive change in patients with Huntington disease on the Repeatable Battery for the Assessment of Neuropsychological Status. J Clin Exp Neuropsychol 2010;32(6):573-8
- Leoni V, Mariotti C, Tabrizi SJ, Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington's disease. Brain 2008;131(Pt 11):2851-9
- Hu Y, Chopra V, Chopra R, Transcriptional modulator H2A histone family, member Y (H2AFY) marks Huntington disease activity in man and mouse. Proc Natl Acad Sci USA 2011;108(41):17141-6
- Dalrymple A, Wild EJ, Joubert R, Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res 2007;6(7):2833-40
- Hardy J, Guerreiro R, Lovestone S. Clusterin as an Alzheimer biomarker. Arch Neurol 2011;68(11):1459-60
- Weiss A, Klein C, Woodman B, Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington's disease. J Neurochem 2008;104(3):846-58
- Weiss A, Abramowski D, Bibel M, Single-step detection of mutant huntingtin in animal and human tissues: a bioassay for Huntington's disease. Anal Biochem 2009;395(1):8-15
- Fang Q, Strand A, Law W, Brain-specific proteins decline in the cerebrospinal fluid of humans with Huntington disease. Mol Cell Proteomics 2009;8(3):451-66
- Constantinescu R, Romer M, Zetterberg H, Increased levels of total tau protein in the cerebrospinal fluid in Huntington's disease. Parkinsonism Relat Disord 2011;17(9):714-15
- Baker K, Queller S, Clough MJ, The Huntington's disease (HD) Toolkit Project; using meta-analysis to identify cognitive tests for use in clinical trials. Clinical Genetics 2011. 80-S1), 47
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010;6(3):131-44