
ABSTRACT
Precision or personalized medicine through clinical genome and exome sequencing has been described by some as a revolution that could transform healthcare delivery, yet it is currently used in only a small fraction of patients, principally for the diagnosis of suspected Mendelian conditions and for targeting cancer treatments. Given the burden of illness in our society, it is of interest to ask how clinical genome and exome sequencing can be constructively integrated more broadly into the routine practice of medicine for the betterment of public health. In November 2014, 46 experts from academia, industry, policy and patient advocacy gathered in a conference sponsored by Illumina, Inc. to discuss this question, share viewpoints and propose recommendations. This perspective summarizes that work and identifies some of the obstacles and opportunities that must be considered in translating advances in genomics more widely into the practice of medicine.
Introduction
Genetic testing is the analysis of human DNA, RNA, or proteins to detect gene variants associated with certain diseases or conditions; non-diagnostic uses include paternity testing and forensics. Genetic testing methodology varies. Molecular genetic tests study single genes or short lengths of DNA to identify variations or mutations that lead to a genetic disorder. Chromosomal genetic tests analyze whole chromosomes or long lengths of DNA to detect large genetic changes such as an extra copy of a chromosome. Finally, biochemical genetic tests study the amount or activity level of proteins; abnormalities in either can indicate changes to the DNA that result in a genetic disorder [Citation1]. summarizes the various applications of genetic testing available today. The last decade has seen an unprecedented pace of advancement in our ability to sequence the genome. As the cost of sequencing decreases, the opportunity to move from targeted sequencing to whole exome sequencing (the analysis of all a person’s genes) and then to whole genome sequencing that analyzes a person’s entire genetic code becomes more accessible, particularly for researchers. This article serves as a high level summary of a Summit meeting in which the implications and obstacles of integrating clinical genome and exome sequencing (CGES) into the practice of medicine.
Table 1. Summary of genetic testing.
The usefulness of CGES in clinical care – clinical utility – is a highly debated topic and this article is intended to supplement a wide-ranging discussion. We encourage readers to explore the literature describing examples of CGES in diagnosing rare conditions[Citation2]; a PubMed search of all English-language articles using the search terms ‘exome’ and ‘utility’ currently returns more than 200 results that describe the use of exome sequencing to reveal conditions such as galactsialidosis [Citation3], ataxia syndromes including Joubert syndrome [Citation4–Citation6], familial hypercholesterolaemia [Citation7], and neurodevelopmental disorders [Citation8]. Among these results our colleagues have explored the utility of exome sequencing in adult and pediatric care and have found that while CGES today is not a substitute for targeted sequencing of suspected genes, positive CGES results can be clinically useful in several ways, particularly among well-selected patients for whom the likelihood for identifying the causative gene change may be as high as 30% [Citation9]. The dialogue on clinical utility of exome sequencing will continue for many years. In our view, which is certainly guided by the research and clinical application of CGES pursued by many of us, clinical utility has been demonstrated, particularly in the oncology field. Broadening utility beyond oncology will take research, resources, and new approaches in regulation.
From advocacy to ‘Free the Data’ to calls for more oversight, the regulation of genetic tests will also continue to evolve. In the United States, three governmental agencies regulate genetic testing. The Centers for Medicare and Medicaid Services (CMS) regulate clinical laboratories, specifically educational requirements for technicians, quality control of laboratory processes, and proficiency testing via the Clinical Laboratory Improvement Amendments of 1988 (CLIA). The US Food and Drug Administration (US FDA) regulates the effectiveness and safety of genetic tests as medical devices under the Medical Devices Amendments of 1976 to the Federal Food, Drug, and Cosmetic Act. Genetic tests can come to market as a commercial test ‘kit’ (a group of reagents packaged and sold to multiple labs) or as a laboratory-developed test (LDT) performed by a single laboratory. FDA has practiced ‘enforcement discretion’ over that latter for many years; however, citing the growing complexity and increasing risk associated with LDTs, FDA has signaled that a shift toward greater oversight is on the horizon [Citation10]. The LDT industry including the lobbying arm of the American Clinical Laboratory Association are investing considerable resources to oppose increased oversight. Their efforts include guiding legislation to reduce the FDA’s authority and oversight of medical testing [Citation11,Citation12]. Several leading opinions have been published to inform this import discussion [Citation13,Citation14].
Additionally, the College of American Pathologists (CAP) has developed a next-generation sequencing (NGS) checklist for CAP-accredited laboratories which they cite as providing ‘another layer of detail’ to CLIA guidelines for NGS [Citation15,Citation16]. Finally, the U.S. Federal Trade Commission (FTC) regulates advertising to ensure that consumers do not receive health-related information that is false or misleading [Citation17].
CGES and cancer care
The Summit focused on the non-oncological application of CGES. CGES has been well-incorporated in cancer prediction and therapy [Citation18–Citation26]. Gagan and Van Allen (2015) provide a current review of high-throughput sequencing in the standard clinical practice of oncology. In addition to assay methodology and recommendations for sample choice, they point to three examples of utility for clinicians: (1) diagnosis; (2) identification of targeted therapy; (3) using NGS in the event a patient stops responding to a targeted therapy with known resistance mutations [Citation24]. The success of sequencing in cancer care stands largely on the shoulders of companion diagnostics. The use of in vitro diagnostics (IVDs) to provide information for the effective and safe use of a corresponding therapeutic product means timely precision cancer therapy. Today’s cancer companion diagnostic market is considerable and growing. illustrates current (as of December 2015) FDA-cleared or approved companion diagnostics and their intended use and indications for use. Furthermore, this success has encouraged a shift in drug development toward pipelines that feature co-development of a test and drug for simultaneous submission to FDA. From this paradigm shift we may bring a market correction of sorts to the therapeutic landscape whereby molecular diagnostics come first followed by assignment to a matching therapy [Citation27].
Table 2. List of FDA-cleared or approved companion diagnostic devices (in vitro and imaging tools).
The pathway for the translation and integration of genomics from the laboratory into everyday medicine remains largely undefined. In November 2014, 46 multidisciplinary stakeholders from academia, industry, clinical delivery, policy, and patient advocacy gathered to assess the challenges of genomic medicine, and to generate recommendations to guide the translation of genomic discoveries into the everyday, non-oncological practice of medicine. The goal of this effort was to assess the current state of clinical applications of genetic and genomic technology, identify the evidence gaps necessary to inform the appropriate utilization of CGES, and propose recommendations that might fill these gaps. Between November 2014 and June 2015, working groups communicated regularly to refine recommendations for what would be needed to facilitate the translation of whole genomic sequencing into everyday, non-oncological practice of medicine. The resulting report identifies scientific and policy areas where additional attention and evidence development are needed, and proposes a number of recommendations, including: (1) generating evidence that quantifies medical utility and cost-effectiveness; (2) customizing regulation to specific use cases; (3) supporting innovative payment models to integrate research and development with clinical care; (4) promoting scalable and iteratively designed ‘learning’ data systems; and (5) educating providers and patients about both the limitations and potential of genomics in medicine. The topic areas and working groups were organized around: (1) clinical utility; (2) regulation; (3) reimbursement; (4) data management; and (5) provider and patient education.
The stakeholders have been described above as ‘multidisciplinary’. The Summit also benefitted from the international perspective that several attendees brought from their clinical practices and patient experiences in Australia, Canada, Italy, and the Republic of Singapore. Nonetheless, the combination of our limited time together in conference and the similarities among the challenges we explored, resulted in a report with a decidedly US-centric perspective. However, we believe that the recommendations and perspectives presented here are not wholly unique to American healthcare but may also be useful abroad.
Clinical utility
At the present time, clinical utility is defined idiosyncratically in the non-oncological space by a diverse range of stakeholders: patients, parents (for pediatric patients), physicians, genetic counselors, hospital administrators, health-plan administrators, and payers. The definition of utility in clinical genetics ranges from definitively informing medical management to produce a positive health outcome, to satisfying the need for patients and their families to have a diagnosis, regardless the outcome. Is it medically useful to identify a molecular diagnosis in a child whose parents have pursued a diagnostic odyssey? [Citation28] In some cases it may be [Citation29]. For those many cases that CGES does not reveal additional medical benefit to the child, objective benefits to the family may be having the opportunity to identify new treatment options, cease costly medical testing that will not prove beneficial, enact reproductive planning for future children, or provide a family with new research opportunities in partnership with others with similar mutations.
However defined, clinical utility is difficult to quantify in genomics, in large part because the current and potential usage of genomics in medicine is so varied. CGES is principally used in assessing the causes of rare, undiagnosed diseases [Citation30], where it is difficult to aggregate patients with the same condition. Emerging categories of genomic sequencing usage include broader pre-conception screening for recessive conditions, non-invasive pre-natal screening (NIPS), pharmacogenomic variation with decision support, and pre-dispositional testing for both Mendelian and common complex disease risk in ostensibly healthy individuals [Citation31]. In each of these examples, there are issues of downstream medical costs and iatrogenic harm that must be considered. In whatever subset of individuals is considered for genomic testing, the probabilistic nature of genetic risk information means that elevated risk will be identified in persons who will never develop the condition, and reduced risk will be identified in persons who will develop the condition. Neither healthcare providers nor patients are skilled at managing probabilistic concepts of risk and only a modest amount of empirical data on utilization or downstream consequences are currently available. Collecting the data to address these questions will require significant research investment and should be high priority for funding agencies. Data collection along these lines is already underway through numerous NIH consortia and projects [Citation32,Citation33], and will be accelerated by the Precision Medicine Initiative [Citation34]. However, defining clinical utility will be a long-term enterprise and the continuing evolution of both technology and medical discovery will mean that such research will continually lag behind the latest discoveries, particularly as discoveries in epigenetics, gene expression, proteomics, and metabolomics are layered upon increasing knowledge about genome sequencing. Providers and patients will thus chronically be in the position of attempting to utilize medical genomics with an under-developed evidence base. This is, to a large extent, inherent in the practice of medicine of all types; for example, whenever providers customize an amalgam of evidence-based knowledge and face-valid guesswork for a given patient, and attempt to communicate uncertainty while simultaneously (and often paradoxically) projecting confidence.
The use of genomics in medicine will exacerbate this issue because genomics brings with it a social narrative of exaggerated determinism. It is also a field of medical science that is changing with particular rapidity and one in which providers feel particularly underprepared [Citation35]. While practitioners can be expected to adjust to genomics as they have to prior technological advances [Citation36], this adjustment may be accelerated by the creation of checklists (or the virtual equivalent embedded in electronic decision support) that reassure providers and patients of the options and uncertainties inherent in genomic medicine. One could envision checklists developed for different types of providers and specialties, for a variety of indications, and attuned to patients and their clinical profiles. This relatively simple solution could be subsequently adapted into broader, more robust educational tools such as pre-counseling videos to expand the knowledge of the patient and provider, and to support their shared decision-making as genomic medicine is integrated into clinical care.
A pre-test checklist could frame communication strategies for providers and to educate patients about genomic testing, including: (1) the analytic and interpretive limitations of the testing so patients are fully informed and can confidently decide if they want to proceed; (2) the potential implications of test results for biological relatives; and (3) the extent of variant data available from a particular test, the wide range of evidence supporting those variants, and the choices and implications of accepting or rejecting that information. A post-test checklist or algorithm, perhaps akin to the American College of Medical Genetics and Genomics ACTion (ACT) sheets available online for the management of rare metabolic conditions [Citation37], could help the provider and patient focus on: (1) what to do if a definitive diagnosis is made; (2) next steps if a potential diagnosis is made; (3) next steps if nothing is found; (4) how to approach secondary findings; (5) how to approach the dynamic nature of the analysis of the patient’s genome; and (6) development of a re-analysis strategy with the patient and/or family members.
The communication of genetic information from provider to patient is an exchange influenced by many variables. Provider readiness and what a clinician deems is relevant to their patient’s case may determine the utility of the genetic test. Similarly, patient expectations of the genetic information may determine how much information they ultimately desire and subsequently receive. Thus, the communication of secondary findings or findings that predict diseases or disorders for which no cure is presently available (non-actionable conditions) must be anticipated by the provider and counseling around these issues provided to the patient and family in each clinical case, whether facilitated by test checklists as described above or by other means.
Finally, as genetic data accumulates within our healthcare system there is a great deal discussion about protecting it from unauthorized access. The topic of privacy protection is discussed in more detail in a later section, but it should be noted that the utility of clinical genomic data will be closely tied to a patient’s confidence in the secure management of their genetic information. Given our continually evolving understanding of genomic information for clinical application we have an opportunity to serve those patients whose data-sharing preferences change over time.
Regulation
Access to medical technologies is regulated by the FDA. Drug, device, and diagnostic manufacturers must submit evidentiary support to FDA demonstrating the safety, efficacy, and quality of their product to receive pre-market approval.
In the area of genomic medicine, the FDA previously adopted a position of enforcement discretion, allowing progress to proceed under the umbrella of LDTs [Citation14]. More recently, the FDA has undergone institutional reorganization and published several guidances [Citation38], and has gradually issued approvals for specific use cases in genomic medicine [Citation39]. However, FDA plans for oversight of LDTs in the realm of genomics are widely regarded as flawed because the FDA may lack statutory authority to regulate LDTs, has an antiquated and unwieldy medical device framework that is not well aligned with the rapidly changing technical and evidence base in genomics, and may inhibit discovery and innovation – in the absence of evidence of harm – by insisting upon pre-market review of both analytic and clinical validity [Citation9,Citation13].
Use cases
Rather than conceptualizing genetic and genomic testing as a device, the output of a genetic test should be regarded as information that can guide clinical decision-making in combination with other patient-specific, non-genetic information at multiple points along the clinical care continuum, from preventive to targeted treatment. In this context, it may be useful to clearly define various ‘use cases’ across that continuum and consider treating them distinctly with highly specific guidelines for the documentation of analytic validity and subsequently, clinical validity. Additionally, given the increasing accessibility and decreasing cost of testing, it is also time to more clearly distinguish genetic information used for ancestry or non-medical traits from genetic information intended for medical use. Certainly some genetic tests may reside in both medical and non-medical categories, such as the personal use of genetic information associated with obesity, exercise, nutrition, and other wellness topics.
Up until the debate around FDA’s jurisdiction over LDTs, the Agency’s position that genetic tests are medical devices has gone largely undisputed. The LDT discussion will stretch into 2016 and the outcome will hinge on whether FDA or the healthcare industry and biomedical research lobbies put forth, and win with, a compelling argument. Today, FDA stratifies medical devices across three classes reflecting their perceived degree of risk. Class I devices pose the lowest level of risk and are subject only to general controls, which include good manufacturing practices, record keeping, and filing specified reports with the agency [Citation40]. Class II devices pose somewhat greater risk and are subject to additional ‘special controls’, such as performance standards, post-market surveillance, patient registries, and device-specific guidances [Citation40]. Class III devices are considered to pose the greatest risk and companies introducing new types of Class III devices must submit an application for pre-market approval (PMA) to the agency [Citation40,Citation41].
Clinical utility as a concept should focus on establishing usefulness and value for patients. Therefore, the demonstration of clinical utility does not, and perhaps cannot, depend on the subjective view of the individual patient. While analytical validity (test is accurate and reliable) and clinical validity (result is medically meaningful) are essentially straightforward metrics, whether the test improves healthcare (clinical utility) is fraught with subjective discordance across the field of medical genetics. Clinical utility as a prerequisite for Medicare coverage is a long-standing and accepted practice; however it struggles align with the vision of personalized medicine. Regardless, demonstrating the clinical utility of any therapeutic or medical device is in the best interest of patient health and the cost of their health management. Precision medicine will simply have to view the burden of evidence gathering as more than convincing regulators and latent adopters and instead approach it from the patient perspective.
There may be many ways to do this, with just one formulation illustrated by where place of use cases into the FDA risk class structure as based on (1) our estimate of the risk/benefit trade-off in these use cases; (2) patient expectation of the test; and (3) alternatives to the use case, e.g. specific test, family history, likely impact on health outcome. By applying varying degrees of risk and benefit to various categories of CGES and placing them into appropriate evidentiary thresholds required to mitigate risk, greater progress may be made in the conversation around regulation.
Figure 1. Defining CGES use cases along the clinical care continuum and appropriate evidentiary thresholds for each.
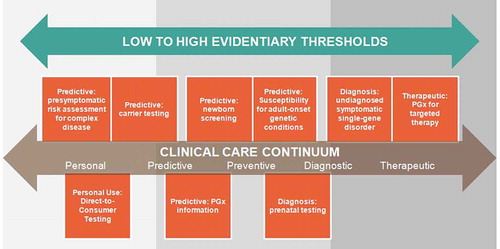
Reimbursement
Reimbursement decisions for new technologies are influenced, but not wholly driven by FDA approval. Several factors are considered for coverage decisions (what gets covered) and reimbursement decisions (how much gets covered), including patient interest/consumer demand; age or pathology-specific applications; inpatient/outpatient status; the opinions of professional organizations; integration into current clinical guidelines; cost-effectiveness; health outcomes; and strength of the evidence supporting a test’s analytical and clinical validity and its clinical utility [Citation42]. Because incontrovertible evidence is almost never available, newer technologies such as genetic testing may be reimbursed inconsistently or not at all. For example, a few years ago, the AlloMap test for cardiac allograft rejection risk (which was not FDA-approved), and a pharmacogenomic test for CYP2C19 effect on clopidogrel (which was FDA-approved) was reviewed by a number of insurers. Two payers provided coverage for these tests, while two different payers considered both tests to be investigational and declined to cover them [Citation42].
Today, genomic testing in the non-oncological space is inconsistently covered throughout the country, which makes it extremely complicated for clinicians to apply such testing in the clinical arena.
It is difficult to predict how this may be resolved, as payers seem to be asking for evidence from clinical utility studies that will take considerable time to accrue. One possibility is that public and private payers could collaborate to expand the use of ‘coverage with evidence development’ programs [Citation43,Citation44]. In this way, data can be collected under research protocols while tests are covered temporarily by third-party payers until sufficient evidence can be gathered to make informed decisions about more permanent coverage. ‘CED programs have been pursued for many years’. Medicare CED programs have demonstrated both success and failure, e.g. pharmacogenomic testing for warfarin response (Decision Memo CAG-00400 N). These performance-based, risk-sharing arrangements where price and level of coverage is linked to clinical effectiveness have proven challenging to implement given their cost and the difficulty of data collection. However, these programs continue to occur globally and Medicare has demonstrated interest in continuing these and similar effort by issuing guidances in 2012 and 2014 that includes a recommendation to partner with the Agency for Healthcare Research and Quality (AHRQ) to conduct CED activities and the specific expectations of a CED study and expected deliverables [Citation45–Citation47].
In recent years a parallel model that focused on genetic testing was fostered by the Medicare contractor Palmetto GBA; the model is called MolDx. For new tests MolDX performs technical assessments to determine whether the test meets coverage requirements. MolDx technical assessments require test developers to submit evidence demonstrating the analytical validity, clinical validity, and clinical utility for each diagnostic test. MolDx will accept a number of different forms of evidence demonstrating clinical utility, each weighted differently: published peer-reviewed articles, RCTs or other well-designed controlled trials, cohort and case study analysis, and articles that are ‘accepted for publication’. Positive assessments are published and coverage determined. If the assessment is negative, MolDx will explain why to the test developer. Workshop attendees at a 2012 NIH-sponsored workshop applauded the Palmetto initiative and discussed whether it could be implemented as a national program to determine coverage for genetic tests. They conceded however that private payers would not be interested in funding similar technical assessments [Citation48].”
Another possibility is for employers to engage more fully with their workforce to support genomic medicine. As employers engage self-funded insurance plans, they have greater incentives to encourage healthy behaviors among their employees; self-insurance also permits employers to customize plans depending on the needs and desires of their workforce [Citation49]. For example, in 2014 more than 70% of employers offering health benefits also provided at least one wellness program, such as weight loss programs, gym membership discounts or on-site exercise facilities, or personal health coaching [Citation50]. Research demonstrating a link between personal genetic risk information in motivating positive health behaviors has not proven promising [Citation51–Citation53]. However, if new evidence continues to accrue demonstrating that genomic testing is indeed useful in promoting healthy behaviors; however [Citation54], employers might encourage conversations between employees and their physicians to explore these options.
As has been noted elsewhere [Citation55,Citation56], the Affordable Care Act allows and encourages employers to tie the cost of insurance plans to participation in wellness programs, while the Genetic Insurance Nondiscrimination Act prohibits employers from learning about genetic information. Thus, this legal misalignment may require particularly creative solutions from employers who seek to integrate genomics information into corporate wellness programs.
Data management
Today’s most promising applications of CGES include NIPS, precision cancer care, diagnosis of rare diseases, and pharmacogenomics; yet, as discussed in the introduction, the collective ability to deliver value from the technology lags far behind the development of the technology itself. Integrating genomics into the everyday practice of medicine will require integrated, scalable, and reliable information systems that resemble other critical information infrastructures, rather than bespoke systems cobbled together from vastly divergent research tools. Furthermore, those systems must present genomic information in the medical record in clinically meaningful ways (e.g. not simply scanned into notes).
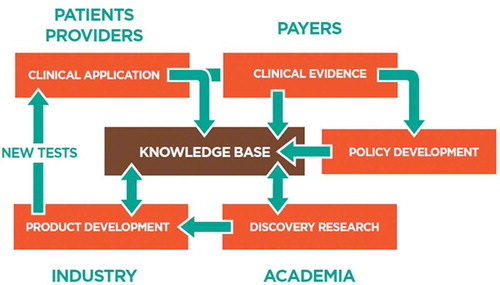
Today’s data management and scalability challenges reflect missing standards, lack of modern data science, and inconsistent security practices, all of which prevent industrial-grade solutions. While genomic data is voluminous, it lacks variety, velocity, and veracity compared to other fields. The number of analysts working with a given dataset is small, as is the number of transactions on any given genome. The data are neither fused with other data types (e.g. clinical or environmental) nor networked with other cases. We must begin to measure the value of each new subsequent genome in the context of the collective understanding of genetics and disease and the predictability of genomic tests, thereby harnessing a ‘network effect’ with the collection of large numbers of genomes linked to clinical data (see ).
Figure 2. Harnessing the network effect of genome sequencing.
There is a real opportunity to construct an environment that creates and sustains virtuous cycles of research, industrial, and clinical activity, as illustrated in . This will require recruiting patients and their data at the point of care to generate a more integrated view of genomic data science and to reinforce the interdependencies between research, product development, and clinical testing. Giving patients the option to donate their data at the point of care will enable these virtuous cycles. An example in the research arena is the National Human Genome Research Institute-funded Clinical Genome Resource (ClinGen) GenomeConnect patient portal that allows patients to securely share and store detailed genotypic and phenotypic information for the advancement of rare disease research [Citation57].
Figure 3. Virtuous cycles possible for genomic data.
The data science of genome interpretation should be rapidly modernized. For example, we should phase out use of exchange formats as data models and text files as data services and move toward state-of-the-art application program interfaces (APIs) for data services, such as those under development by the Global Alliance for Genomics and Health [Citation58]. There is an urgent need to improve feature extraction from clinical data and to fuse these data with other data, such as environmental and patient-generated data, including data from wearable sensors that automatically collect data about behaviors and the environment. Those data can then be made available to analysts and algorithms in an event-driven manner to promote dynamic analytics and reporting.
There is also a need to modernize the governance of genomic interpretation systems. In many data information systems that serve as critical infrastructures, there is a core, regulated production system as well as a separate development tier that allows experimentation and innovation. Between these two system tiers is a change management process that governs which experimental features are elevated and implemented into the production environment. This type of ‘dev-ops’ approach allows for standardization at the production level, but does not inhibit experimentation. The change management process is an area where regulators can have a voice in what is delivered clinically to patients.
Finally, as many have argued, there is an urgent need to eliminate data silos and promote data sharing [Citation57]. Shifting away from the notion of institutional data ownership (including provider, industry, and academic) to the reality of individual data ownership and engaging patients as partners in research is essential to creating the incentives that will support a sustainable data enterprise for genomic medicine. Companies such as PatientsLikeMe promote sharing models in the hope of accelerating research. Indeed, PatientsLikeMe has signed sharing agreements with FDA and AstraZeneca to advance research in drug safety, lupus, diabetes, and other areas.
Of equal importance is the need to instill a considerable measure of confidence and trust in the data systems we build. We live in an era of data breaches and stolen identities that can cause tremendous financial and reputational losses. The protection of genomic data is quickly emerging as its own research field, with academic thought being invested to balance the need for genomic data with the need for retaining one’s autonomy, what Erlich et al. describe as ‘a solution in which researchers and participants both win’ [Citation59]. Genealogical triangulation, exploiting meta-data, and identity tracing by phenotypic prediction are just some of the methods that can be employed to compromise genomic data sharing by subjects [Citation60,Citation61]. Despite several existing layers of protections, including GINA and HIPAA in the United States, the European Union privacy directive (Directive 95/46/EC, 1995), and the use of institutional review boards and data access committees in research, the pace and increasing size of datasets makes data protection challenging. Coupled with solutions such as homomorphic encryption [Citation62], providers and researchers must clearly inform their patients and subjects about the potential privacy risks when sharing genetic and genomic data.
Provider and patient education
Why are today’s healthcare providers not more rapidly incorporating CGES into their practice of medicine? In some cases, it is because they are unaware of new genomic tools and tests, and when they are aware, many lack self-confidence about their own knowledge and skills for using them [Citation35,Citation63]. Many providers are also concerned about the high cost of genomic tests, the lack of reimbursement, and skepticism about the validity and utility of the tests currently being offered [Citation64]. As noted earlier, a robust genetics education for providers will be essential to address these challenges. Moreover, professional guidelines around genomics must be adopted. Overcoming these challenges will only come as the case is made for the clinical utility and cost effectiveness of genomics in patient care. In the meantime, the teaching of medicine should reject the historical exceptionalism that has surrounded genomics, present genetics as foundational in the basic science years, and model the practice of genomic medicine at every opportunity during clinical training. At the same time educational programs should include broader historical perspectives on genetics in medicine and society. The troubled history of the human genetics in the first half of the twentieth century directs us toward caution and vigilance.
It has also been suggested that trainees or practicing providers who obtain genetic testing themselves may have enhanced educational engagement around genomics, and many medical schools are considering incorporating such programs into their genetics curricula [Citation65]. This fits well with the traditional ‘learning by doing’ physician education model. However, pilot projects that have explored such programs have generated mixed results [Citation63,Citation66–Citation69], and at least one institution considered and rejected the idea due to concerns about coercion, privacy, conflicts of interest, and safety monitoring [Citation70].
‘Indeed, asking medical students to undergo genetic testing in order to learn about genomics could be considered a form of genetic exceptionalism. Furthermore, approaching a medical student to request that they be unnecessarily treated with a medical technology or therapeutic is inappropriate.’
At the same time, patients are playing a more substantive role in their own health care and their engagement with precision medicine in general, and genomics specifically, may shape how these concepts are realized. Patients are increasingly seeking ways to empower themselves and their families with information obtained outside of interactions with their healthcare practitioners, and many are embracing self-monitoring systems that measure physical activity, sleep patterns, weight, and even blood chemistry [Citation71,Citation72]. At present, only a small fraction are taking advantage of relatively new opportunities to access their medical records [Citation73,Citation74], but patient portals that take two-way communications beyond appointment setting and billing and into clinical discussions may present new, appropriate opportunities to increase engagement of patients with genomics. Beyond these steps, there is an expectation that patients will increasingly share their experiences, their clinical data, and even their genomic data among themselves and with researchers. Sharing is hampered; however, by technological limitations, legal protections, and social issues. Recent legislation (Title 42 of the Code of Federal Regulations (CFR) Public Health, Part 493) requiring laboratories to make test results available to consumers is a positive step toward personal data stewardship, but it is unclear whether that right would extend to genomic data, particularly unanalyzed (raw) data. A critical part of advancing genomic aspects of precision medicine is to educate and engage not only providers, but consumers of health care as well. Providing patients/consumers access to their health data coupled with high-quality educational materials is the first step.
Expert commentary
The Summit and subsequent conference calls of the respective working groups offered a productive opportunity to gather stakeholders from multiple disciplines to compare and contrast views on the current challenges to the integration of clinical genomics into the practice of medicine. At the close of the conference attendees were asked to estimate the number of years until such time that CGES is fully integrated into the care of most Americans; the overall consensus was 8 to 10 years. It remains to be seen whether this is sufficient time to accrue the evidence for clinical utility, revise and build a nimble and appropriate regulatory and policy framework, stand up a payment system that fosters medical innovation, unleash the power of large datasets, and sufficiently educate providers and patients.
The time-frame for integration is no longer predicated on scientific development; rather integration depends on concerted efforts among and between the healthcare stakeholders in precision medicine. Providers, payers, and patients along with policy makers and regulators, industry and academia share the expertise and interest to push toward clinical care powered in part by genetics and genomics. illustrates our recommendations juxtaposed with these stakeholders to demonstrate where they can collaborate on activities that support these and similar recommendations.
Figure 4. Stakeholders collaborating to catalyze precision medicine.
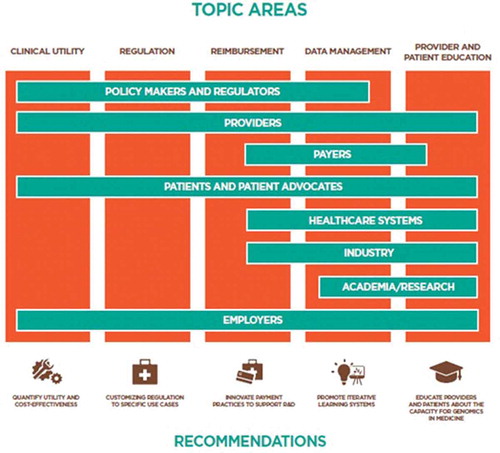
This figure is not exhaustive. There may be broader opportunity for collaboration among stakeholders on these recommendations, for example, academia can certainly contribute to the demonstration of utility in a variety of ways, including the study of economic benefit of CGES. The principal purpose of this figure is to illustrate that cross-sector cooperation is imperative to expanding the use of CGES in medicine at a useful pace.
Five-year view
The momentum of several efforts, including the Precision Medicine Initiative, has opened a window of opportunity to integrate genomic medicine into the clinical setting. If these efforts can be complemented by well-orchestrated, collaborative efforts by a range of stakeholders pursuing activities designed to accelerate a shared vision of precision medicine, significant gains in genome-informed clinical care may be feasible by the close of this decade. Healthcare systems, providers and payers, employers and patient consumers, and industry should coalesce around meaningful opportunities such as patient education, concordance in coverage policy, and clinical guidelines to promote wider adoption.
The last decade saw great strides in our understanding of disease and pharmacogenomics. It also marked the movement of genetics and genomics into mainstream vernacular. It is our hope that the next decade of precision medicine will be characterized by equal parts progress in discovery, technology, and clinical implementation. This progress will be measured by demonstrated access to CGES across different populations as we move from affluent early adopters to patients often underserved by the healthcare system, and improved health outcomes.
Key issues
The pathway for the translation and integration of genomics from the laboratory into everyday medicine remains largely undefined.
The integration of whole genomic sequencing into everyday, non-oncological practice of medicine requires additional attention and evidence development in key scientific and policy areas: (1) generating evidence that quantifies medical utility and cost-effectiveness; (2) customizing regulation to specific use cases; (3) supporting innovative payment models to integrate research and development with clinical care; (4) promoting scalable and iteratively designed ‘learning’ data systems; and (5) educating providers and patients about both the limitations and potential of genomics in medicine.
Orchestrated efforts by a range of stakeholders, namely healthcare systems, providers and payers, employers, patient consumers and advocates, policy makers and regulators, academia, and industry can accelerate a shared vision of precision medicine by pursuing collaborative activities in provider and patient education, data generation and broad sharing practices, developing concordance in regulatory and reimbursement policy, and utility studies focused toward informing genomic clinical guidelines.
Financial and competing interests disclosure
Speakers were compensated by Illumina, Inc. for their travel to the conference and in some cases were paid an honorarium for speaking at the conference, but were not compensated for any work on this paper.
SK Delaney and MF Christman are supported by United States Air Force cooperative agreement FA8650-14-2-6533. JW Belmont is supported by NIH/NHLBI 5RO1 HL090506. J Quackenbush is supported by NIH/NHLBI 5P01 HL105339 and NCI 2P50 CA127003. TS Hays is supported by NIH/NIGMS R01 GM44757. RC Green is supported by NIH grants U01-HG006500, U19-HD077671, R01-HG005092, U01-HG008685 and U41-HG006834, as well as funding from the US Department of Defense.
M Delledonne is co-founder of Personal Genomics s.r.l., a genomic interpretation company and spin-off of University of Verona, Italy.
JR Friedman has family investments in the biotechnology/genomics space.
RC Green’s work has been funded by the NIH, Department of Defense and Illumina, Inc. He has also received compensation for consulting with Bina, Prudential, Invitae, Illumina, Arivale and Helix.
J Huber serves on the board of Illumina, Inc.
N Ledbetter is a consultant to Natera, Inc.
W H Lee is a consultant to Life Letters, a company offering genetic testing in Australia and CNSDose which is commercialising 2nd generation pharmacogenetic guidance for selection and dosage of anti-depressants.
E Levin serves on the Advisory Board of Pathway Genomics, Inc., and on the Scientific Advisory Board of LifeMap Solutions, Inc.
RL Love serves on the Board of Managers for PMed Management LLC.
JJ McCarthy is a consultant for Omicia, Inc.
RL Nussbaum is a full-time employee of Invitae Corp., a genetic testing company.
BA Patay is the Chief Medical Officer of Genection, a company associated with Invivoscribe.
J Quackenbush is co-Founder and Board Chair of GenoSpace, a precision medicine software company. J Quackenbush is a member of the scientific advisory boards of Caris Life Sciences and Perthera.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Acknowledgement
Authors are grateful to Shervin Kamkar, Laura Wachtmann, and Dawn Barry (all of Illumina, Inc.) for providing a thoughtful and structured opportunity to come together and fully explore the landscape.
References
- What Is Genetic Testing? Genetics Home Reference. Bethesda (MD): NIH/U.S. National Library of Medicine 2016. [cited 2016 Jan 16]. Available from: http://ghr.nlm.nih.gov/handbook/testing/genetictesting
- Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Eng J Med. 2014;371(12):1170.
- Prada CE, Gonzaga-Jauregui C, Tannenbaum R, et al. Clinical utility of whole-exome sequencing in rare diseases: galactosialidosis. Eur J Med Genet. 2014;57(7):339–344.
- Tsurusaki Y, Kobayashi Y, Hisano M, et al. The diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J of Hum Genet. 2013;58(2):113–115.
- Pyle A, Smertenko T, Bargiela D, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain. 2015;138(2):276–283.
- Tacik P, Guthrie KJ, Strongosky AJ, et al. Whole-exome sequencing as a diagnostic tool in a family with episodic ataxia type 1. Mayo Clin Proc. 2015;90(3):366–371.
- Futema M, Plagnol V, Whittall RA, et al. Use of targeted exome sequencing as a diagnostic tool for familial hypercholesterolaemia. J Med Genet. 2012;49(10):644–649.
- Dixon-Salazar TJ, Silhavy JL, Udpa N, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Trans Med. 2012;4(138):138ra178–138ra178.
- Biesecker LG, Biesecker BB. An approach to pediatric exome and genome sequencing. Curr Opin Pediatr. 2014;26(6):639–645.
- FDA. The public health evidence for FDA oversight of laboratory developed tests: 20 case studies. Silver Spring (MD): FDA; 2015.
- The Complex Effects of the FDA’s Proposal to Regulate Laboratory-Developed Tests. Sachs R. HealthAffairs Blog 2015. [Last accessed 2016 Jan 14]. Available from: http://healthaffairs.org/blog/2015/04/10/the-complex-effects-of-the-fdas-proposal-to-regulate-laboratory-developed-tests/.
- Is Lab Testing the ‘Wild West’ of Medicine? Burton TM. Wall Street Journal 2015. [cited 2016 Jan 14]. Available from: http://www.wsj.com/articles/is-lab-testing-the-wild-west-of-medicine-1449800707.
- Evans JP, Watson MS. Genetic testing and FDA regulation: overregulation threatens the emergence of genomic medicine. JAMA. 2015;313(7):669.
- Evans BJ, Burke W, Jarvik GP. The FDA and genomic tests–getting regulation right. N Engl J Med. 2015;372(23):2258–2264.
- Heger M. CAP publishes accreditation checklist for NGS in clinical labs [Internet]. GenomeWeb. 2012 [cited 2016 Jan 8]. Available from: https://www.genomeweb.com/sequencing/cap-publishes-accreditation-checklist-ngs-clinical-labs)
- Aziz N, Zhao Q, Bry L, et al. College of American Pathologists’ laboratory standards for next-generation sequencing clinical tests. Arch Pathol Lab Med. 2015;139(4):481–493.
- Division of Advertising Practices. Washington, DC: FTC/Bureau of Consumer Protection 2016. Available at: https://www.ftc.gov/about-ftc/bureaus-offices/bureau-consumer-protection/our-divisions/division-advertising-practices [Last accessed 14 January 2016].
- Lerman C, Narod S, Schulman K, et al. BRCA1 testing in families with hereditary breast-ovarian cancer. A prospective study of patient decision making and outcomes. JAMA. 1996;275(24):1885–1892.
- Jiang Y, Wang M. Personalized medicine in oncology: tailoring the right drug to the right patient. Biomark Med. 2010;4(4):523–533.
- Verma M. Personalized medicine and cancer. J Personalized Med. 2012;2(1):1–14.
- Walko CM, McLeod H. Use of CYP2D6 genotyping in practice: tamoxifen dose adjustment. Pharmacogenomics. 2012;13(6):691–697.
- Vijayaraghavan A, Efrusy MB, Göke B, et al. Cost-effectiveness of KRAS testing in metastatic colorectal cancer patients in the United States and Germany. Int J Cancer. 2012;131(2):438–445.
- Weng L, Zhang L, Peng Y, et al. Pharmacogenetics and pharmacogenomics: a bridge to individualized cancer therapy. Pharmacogenomics. 2013;14(3):315–324.
- Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome Med. 2015;7(1):80.
- Tripathy D, Harnden K, Blackwell K, et al. Next generation sequencing and tumor mutation profiling: are we ready for routine use in the oncology clinic? BMC Med. 2014;12:140.
- Johnson DB, Dahlman KH, Knol J, et al. Enabling a genetically informed approach to cancer medicine: a retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next-generation sequencing panel. The Oncologist. 2014;19(6):616–622.
- Illumina to Support ‘Companion Therapeutics’ in Oncology. Bio-IT World. Needham, MA 2014. [cited 2016 Jan 14]. Available from: http://www.bio-itworld.com/2014/8/21/illumina-support-companion-therapeutics-oncology.html.
- Carmichael N, Tsipis J, Windmueller G, et al. “Is it going to hurt?”: the impact of the diagnostic odyssey on children and their families. J Genet Couns. 2015;24(2):325–335.
- American College of Medical Genetics and Genomics. Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2015;17(6):505–507.
- Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014;370:2418–2425.
- Green RC, Rehm HL, Kohane IS. Clinical genome sequencing. In: Ginsburg GS, Willard HF, editors. Genomic and personalized medicine. San Diego (CA): Academic Press; 2013. p. 102–122.
- Genomic Medicine Activities. NIH/NHGRI/Division of Genomic Medicine. Bethesda, MD 2015. [Last accessed 2016 Feb 14]. Available from: http://www.genome.gov/27549225.
- Current Research Programs. NIH/NHGRI/Division of Genomic. Bethesda, MD 2015. [cited 2016 Feb 14]. Available from: http://www.genome.gov/27551170.
- Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–795.
- Powell KP, Cogswell WA, Christianson CA, et al. Primary care physicians’ awareness, experience and opinions of direct-to-consumer genetic testing. J Genet Couns. 2012;21(1):113–126.
- Vassy JL, Korf BR, Green RC. How to know when physicians are ready for genomic medicine. Sci Trans Med. 2015;7(287):287fs19–287fs19.
- ACT Sheets and Confirmatory Algorithms. American College of Medical Genetics and Genomics. Bethesda, MD, 2016. [cited 2016 Jan 14]. Available from: https://www.acmg.net/ACMG/Publications/ACT_Sheets_and_Confirmatory_Algorithms/ACMG/Publications/ACT_Sheets_and_Confirmatory_Algorithms/ACT_sheets_Homepage.aspx?hkey=6d43e3d3-71fd-49f4-88d5-7197238f9f33.
- Paving the way for personalized medicine. FDA. Silver Spring, MD, 2013. [cited 2015 Dec]. Available from: http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/PersonalizedMedicine/UCM372421.pdf.
- Collins FS, Hamburg MA. First FDA authorization for next-generation sequencer. N Engl J Med. 2013;369(25):2369–2371.
- Classification of devices intended for human use (c). United States Code, 2006 Edition, Supplement 4, Title 21 – Food and Drugs. [cited 2016 Jan 14]. Available from: https://www.gpo.gov/fdsys/granule/USCODE-2010-title21/USCODE-2010-title21-chap9-subchapV-partA-sec360c/content-detail.html.
- Javitt GH, Carner KS. Regulation of next generation sequencing. J Law Med Ethics. 2014;42 Suppl 1:9–21.
- Hresko A, Haga SB. Insurance coverage policies for personalized medicine. J Pers Med. 2012;2(4):201–216.
- Tunis SR, Pearson SD. Coverage options for promising technologies: Medicare’s ‘coverage with evidence development’. Health Aff. 2006;25(5):1218–1230.
- Khoury M, Berg A, Coates R, et al. The evidence dilemma in genomic medicine. Health Aff. 2008;27(6):1600–1611.
- Mei Li C, Risebrough, Nancy, Hux Marg Coverage with evidence development activities around the world: an environment scan. ISPOR 17th Annual European Congress. Amsterdam, The Netherlands, 2014
- Medicare’s Reset on ‘Coverage With Evidence Development. Neumann PJ, Chambers J. HealthAffairs Blog. 2013; [cited 2016 Jan 14]. Available from: http://healthaffairs.org/blog/2013/04/01/medicares-reset-on-coverage-with-evidence-development/.
- Coverage with Evidence Development. Guidance for the Public, Industry. CMS/Medicare. Baltimore, MD. 2014. [cited 2016 Jan 14]. Available from: https://www.cms.gov/Medicare/Coverage/Coverage-with-Evidence-Development/.
- Reimbursment models to promote evidence generation and innovation for genomic tests. NIH. Bethesda (MD). 2012; [cited 2016 Jan 14]. Available from: https://www.genome.gov/27552210.
- Self-insured Group Health Plans: questions and answers. Self-Insurance Institute of America. Washington, DC. 2015. [cited 2016 Jan 14]. Available from: http://www.siia.org/i4a/pages/Index.cfm?pageID=4546.
- Employer Health Benefits 2014 Annual Survey. Claxton G, Rae M, Panchal N et al. The Henry J. Kaiser Family Foundation and Health Research & Educational Trust. Menlo Park (CA). 2014; [cited 14 January 2016]. Available from: https://kaiserfamilyfoundation.files.wordpress.com/2013/04/8225.pdf
- Henrikson NB, Bowen D, Burke W. Does genomic risk information motivate people to change their behavior? Genome Med. 2009;1(4):37–37.
- Bloss CS, Madlensky L, Schork NJ, et al. Genomic information as a behavioral health intervention: can it work? Per Med. 2011;8(6):659–667.
- Webster TH, Beal SJ, Brother KB. Motivation in the age of genomics: why genetic findings of disease susceptibility might not motivate behavior change. Life Sciences, Society Policy. 2013;9:8.
- Diseati L, Scheinfeldt LB, Kasper RS, et al. Common genetic risk for melanoma encourages preventive behavior change. J Pers Med. 2015;5(1):36–49.
- Bard JS. When public health and genetic privacy collide: positive and normative theories explaining how ACA’s expansion of corporate wellness programs conflicts with GINA’s privacy rules. J Law Med Ethics. 2011;39:469–487.
- Green RC, Lautenbach D, McGuire AL. GINA, genetic discrimination, and genomic medicine. N Engl J Med. 2015;372(5):397–399.
- Rehm HL, Berg JS, Brooks LD, et al. ClinGen – the clinical genome resource. N Engl J Med. 2015;372(23):2235–2242.
- The Global Alliance for Genomics and Health. Ontario, Canada 2015. [cited 2016 Jan 16]. Available from: https://genomicsandhealth.org/
- Erlich Y, Williams JB, Glazer D, et al. Redefining genomic privacy: trust and empowerment. Plos Biology. 2014;12(11):e1001983.
- Gymrek M, McGuire AL, Golan D, et al. Identifying personal genomes by surname inference. Science. 2013;339(6117):321–324.
- Erlich Y, Narayanan A. Routes for breaching and protecting genetic privacy. Nat Rev Genet. 2014;15(6):409–421.
- Frederick R. Core concept: homomorphic encryption. Proc Natl Acad Sci USA. 2015;112(28):8515–8516.
- Haga SB, Carrig MM, O’Daniel JM, et al. Genomic risk profiling: attitudes and use in personal and clinical care of primary care physicians who offer risk profiling. J Gen Intern Med. 2011;26(8):834–840.
- Manolio TA, Chisholm RL, Ozenberger B, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med. 2013;15(4):258–267.
- Demmer LA, Waggoner DJ. Professional medical education and genomics. Annu Rev Genomics Hum Genet. 2014;15:507–516.
- Sharp RR, Goldlust ME, Eng C. Addressing gaps in physician education using personal genomic testing. Genet Med. 2011;13(8):750–751.
- Ormond KE, Hudgins L, Ladd JM, et al. Medical and graduate students’ attitudes toward personal genomics. Genet Med. 2011;13(5):400–408.
- Salari K, Karczewski KJ, Hudgins L, et al. Evidence that personal genome testing enhances student learning in a course on genomics and personalized medicine. PLoS One. 2013;8(7):e68853.
- Sanderson SC, Linderman MD, Kasarskis A, et al. Informed decision-making among students analyzing their personal genomes on a whole genome sequencing course: a longitudinal cohort study. Genome Med. 2013;5(12):113.
- Walt DR, Kuhlik A, Epstein SK, et al. Lessons learned from the introduction of personalized genotyping into a medical school curriculum. Genet Med. 2011;13(1):63–66.
- Consumer Survey on Patient Engagement: United States Research Recap. Accenture. Chicago, IL. 2013. [cited 2016 Jan 15]. Available from: https://www.accenture.com/t20150708T033734__w__/us-en/_acnmedia/Accenture/Conversion-Assets/DotCom/Documents/Global/PDF/Industries_11/Accenture-Consumer-Patient-Engagement-Survey-US-Report.pdf
- Topol EJ. The creative destruction of medicine: how the digital revolution will create better health care. Basic books. New York (NY): A Member of the Perseus Books Group; 2012.
- Patient Engagement: the Holy Grail of Meaningful Use. peer60/Patient Portals. American Fork, UT. 2014; [cited 2016 Jan 15]. Available from: http://research.peer60.com/patient-portals/
- Wachter R. The digital doctor: hope, hype, and harm at the dawn of medicine’s computer age. Boston (MA): McGraw-Hill Education; 2015.