

Dilated cardiomyopathy (DCM) is a condition characterized by unexplained left ventricle dilatation, impaired systolic function and nonspecific histologic abnormalities dominated by myocardial fibrosis Citation[1–3]. The condition has an estimated prevalence of 36.5 out of 100,000 in the US population, and is the most common cause of heart failure and cardiac transplantation in the young Citation[4,5]. Patients may experience severe disease complications including arrhythmia, thromboembolic events and sudden death due to ventricular arrhythmia.
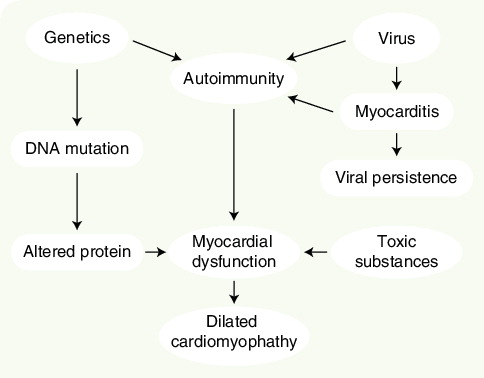
Recent developments in medical treatment have diminished symptoms and improved prognosis considerably [6–10]. In addition, novel biventricular pacing modalities appear promising in the treatment of patients with severe heart failure Citation[11–14]. Despite the fairly uniform clinical expression of DCM, the etiology is poorly understood and appears to be very heterogeneous . Viral infections, autoimmune disease and toxic substances are believed to be causative in a proportion of DCM cases, although definitive proof has often been difficult to obtain [2,15]. Recent studies have suggested that genetic factors may account for as many as 30–50% of cases Citation[16,17]. A large number of disease genes have been identified that encode proteins involved in a variety of cell functions, ranging from force generation within the sarcomere to regulation of myocyte ion channels [18–35]. Most affected families present with a ‘pure’ cardiac phenotype and, in these families, autosomal dominant transmission is most frequently followed by recessive and X-linked inheritance. In addition to impaired cardiac function, variable degrees of skeletal muscle dystrophy and cardiac conduction disease have been reported with mutations in genes such as dystrophin, desmin and emerin. Less frequently, affected individuals present with involvement of other organ systems, such as in Barth syndrome, which is characterized by DCM, neutropenia, abnormal mitochondrial function as well as skeletal myopathy Citation[36,37]. Disease-responsible mutations for this condition have been identified in the gene G4.5, which encodes the protein taffazin and in which mutations may also lead to pure DCM, endocardial fibrosis or left ventricle noncompaction without any features of Barth syndrome. Mitochondrial mutations may be suspected in DCM patients with neurologic deficits and skeletal muscle involvement in addition to symptoms from other organ systems Citation[38].
The results of previous drug trials of heart-failure patients have indicated a beneficial effect of prophylactic pharmacologic therapy in asymptomatic individuals with impaired systolic function Citation[6,9]. Therefore, it is reasonable to assume that early diagnosis and treatment of asymptomatic patients with hereditary DCM may also improve their prognosis Citation[8]. Genetic diagnosis would be helpful in identifying unaffected mutation carriers who require follow-up and would enable termination of clinical screening of relatives with a normal genotype. The potential of genetic diagnosis in clinical management of DCM is large, but, at present, only a few studies have investigated the frequency and clinical expression of recognized DCM genes in large patient cohorts. More research is required to clarify the clinical implication of genetic diagnosis in DCM.
The discovery of novel DCM genes has provided valuable new insight into the potential pathophysiologic basis of the condition. However, the variable disease expression observed, even in families affected by identical mutations, suggests that environmental factors, as well as the entire genetic constitution of individual patients, influence disease expression. A variety of pathways modifying the phenotype are likely to be involved in disease development, necessitating detailed analysis of the failing myocardium from affected individuals using multiple methods including proteomic, microarray and metabolic analyses. Each patient is the product of a complex set of genetic variations, different degrees of influence of diets, lifestyles and drug therapy. The genomic profile of any single patient can be assessed using gene arrays (complementary [c]DNA or oligonucleotide based). The proteins expressed by failing hearts can be investigated using 2D polyacrylamide gel electrophoresis (PAGE), which can resolve several thousand proteins of the most abundant proteins in a single sample. 2D-PAGE struggles to identify proteins expressed at low levels. Technical advances in 2D-PAGE and mass spectroscopy may solve some of these issues. A combined approach integrating microarray, proteomic, metabolite analysis and detailed clinical investigations is the most promising way towards understanding the subtleties of heart failure. Proteomic approaches are valuable in that they can identify changes in protein isoform expression, and even changes in phosphorylation state. Other post-translational modifications, such as glycosylation, can also be detected. Identifying these subtle changes allows the investigator to examine or identify signaling pathways involved in disease progression. This approach may also lead to the identification of pathways not previously known to be involved in DCM. Microarray analysis provides entirely different data that may not always correlate to the proteomic data, since changes in gene expression, evident at the level of mRNA, do not always mean changes in protein levels. These data may be more useful in providing a fingerprint or pattern that identifies a particular disease state. Microarrays are also customizable and easily adapted to high-throughput approaches, so it may be possible to identify a subset of genes that have altered expression in DCM, and use these on a chip to quickly assess or predict a patient’s disease state. This would be especially useful in a clinical setting and as part of a more sensitive phenotypic assessment of otherwise healthy gene carriers.
Examination of the metabolome (metabolite analysis) also provides detailed information about the state of a patient’s heart. Certain disease states (for instance, ischemic heart failure) are associated with marked shifts in metabolic profiles. Usually, the heart uses fatty acids to generate ATP, but glucose utilization for ATP production increases as heart failure progresses. It is relatively easy to assess levels of metabolic intermediates, such as fumarate, succinate, creatine, phosphocreatine, reduced NADH, ATP, ADP and amino acids, to create a metabolic profile for each patient. This allows an assessment of which substrates (fatty acids or glucose) are being used for energy production, and to what extent those substrates are being utilized. This will provide extra information for characterizing the disease state or stage of individual patients.
All of these approaches provide a large amount of information. One of the biggest challenges will first be to identify which subsets of information are useful, and then how to integrate the data to provide meaningful and useful information for the clinician and research scientist alike. Bioinformatics is the key to integrating these data, but it is also the weak point in many scientists’ and clinicians’ training. To take advantage of all of these data, a good working knowledge of bioinformatics is needed.
No studies have yet combined all these techniques to investigate cardiac disease, but some have used either proteomic or microarray approaches to investigate patients or animal models of heart failure. It is striking that each study group (either DCM, ischemic or pressure overload heart failure) has different patters of gene and/or protein expression Citation[39,40]. It will be interesting to see how the proteomes and gene expression profiles of patients with the same disease mutation, but with phenotypically different disease, are altered. It was once believed that there was a final common pathway for heart failure, but it is more likely that there are multiple pathways to a common heart-failure phenotype.
References
- Richardson P, McKenna W, Bristow M et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 93, 841–842 (1996).
- Towbin JA, Bowles NE. The failing heart. Nature 415, 227–233 (2002).
- Mestroni L, Maisch B, McKenna WJ et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur. Heart J. 20, 93–102 (1999).
- Codd MB, Sugrue DD, Gersh BJ, Melton LJ. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 80, 564–572 (1989).
- Cohn JN, Bristow MR, Chien KR et al. Report of the National Heart, Lung, and Blood Institute Special Emphasis Panel on Heart Failure Research. Circulation 95, 766–770 (1997).
- The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N. Engl. J. Med. 327, 685–691 (1992).
- Pfeffer MA, Braunwald E, Moye LA et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N. Engl. J. Med. 327, 669–677 (1992).
- Waagstein F, Bristow MR, Swedberg K et al. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Metoprolol in Dilated Cardiomyopathy (MDC) Trial Study Group. Lancet 342, 1441–1446 (1993).
- Poole-Wilson PA, Swedberg K, Cleland JG et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet 362, 7–13 (2003).
- Cleland JG, Pennell DJ, Ray SG et al. Myocardial viability as a determinant of the ejection fraction response to carvedilol in patients with heart failure (CHRISTMAS trial): randomised controlled trial. Lancet 362, 14–21 (2003).
- Molhoek SG, Bax JJ, van Erven L et al. Comparison of benefits from cardiac resynchronization therapy in patients with ischemic cardiomyopathy versus idiopathic dilated cardiomyopathy. Am. J. Cardiol. 93, 860–863 (2004).
- Mortensen PT, Sogaard P, Mansour H et al. Sequential biventricular pacing: evaluation of safety and efficacy. Pacing Clin. Electrophysiol. 27, 339–345 (2004).
- Blanc JJ, Bertault-Valls V, Fatemi M, Gilard M, Pennec PY, Etienne Y. Midterm benefits of left univentricular pacing in patients with congestive heart failure. Circulation 109, 1741–1744 (2004).
- Auricchio A, Stellbrink C, Butter C et al. Clinical efficacy of cardiac resynchronization therapy using left ventricular pacing in heart failure patients stratified by severity of ventricular conduction delay. J. Am. Coll. Cardiol. 42, 2109–2116 (2003).
- Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104, 557–567 (2001).
- Mogensen J, Murphy RT, Shaw T et al. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 44, 2033–2040 (2004).
- Murphy RT, Mogensen J, Shaw A, Kubo T, Hughes S, McKenna WJ. Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy. Lancet 363, 371–372 (2004).
- Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 280, 750–752 (1998).
- Olson TM, Kishimoto NY, Whitby FG, Michels VV. Mutations that alter the surface charge of α-tropomyosin are associated with dilated cardiomyopathy. J. Mol. Cell Cardiol. 33, 723–732 (2001).
- Kamisago M, Sharma SD, DePalma SR et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 343, 1688–1696 (2000).
- Gerull B, Gramlich M, Atherton J et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nature Genet. 30, 201–204 (2002).
- Itoh-Satoh M, Hayashi T, Nishi H et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 291, 385–393 (2002).
- Daehmlow S, Erdmann J, Knueppel T et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 298, 116–120 (2002).
- Knoll R, Hoshijima M, Hoffman HM et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111, 943–955 (2002).
- Fatkin D, MacRae C, Sasaki T et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 341, 1715–1724 (1897).
- Bione S, Maestrini E, Rivella S et al. Identification of a novel X-linked gene responsible for Emery–Dreifuss muscular dystrophy. Nature Genet. 8, 323–327 (1994).
- Li D, Tapscoft T, Gonzalez O et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 100, 461–464 (1897).
- Tsubata S, Bowles KR, Vatta M et al. Mutations in the human δ-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J. Clin. Invest. 106, 655–662 (2000).
- Muntoni F, Cau M, Ganau A et al. Brief report: deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N. Engl. J. Med. 329, 921–925 (1993).
- Norgett EE, Hatsell SJ, Carvajal-Huerta L et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 9, 2761–2766 (2000).
- Olson TM, Illenberger S, Kishimoto NY, Huttelmaier S, Keating MT, Jockusch BM. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation 105, 431–437 (2002).
- Mohapatra B, Jimenez S, Lin JH et al. Mutations in the muscle LIM protein and α-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol. Genet. Metab. 80, 207–215 (2003).
- Vatta M, Mohapatra B, Jimenez S et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J. Am. Coll. Cardiol. 42, 2014–2027 (2003).
- Schmitt JP, Kamisago M, Asahi M et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299, 1410–1413 (2003).
- Bienengraeber M, Olson TM, Selivanov VA et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nature Genet. 36, 382–387 (2004).
- Ichida F, Tsubata S, Bowles KR et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 103, 1256–1263 (2001).
- Chen R, Tsuji T, Ichida F et al. Mutation analysis of the G4.5 gene in patients with isolated left ventricular noncompaction. Mol. Genet. Metab. 77, 319–325 (2002).
- Suomalainen A, Paetau A, Leinonen H, Majander A, Peltonen L, Somer H. Inherited idiopathic dilated cardiomyopathy with multiple deletions of mitochondrial DNA. Lancet 340, 1319–1320 (1992).
- Barrans JD, Allen PD, Stamatiou D, Dzau VJ, Liew CC. Global gene expression profiling of end stage dilated cardiomyopathy using a human cardiovascular-based cDNA microarray. Am. J. Pathol. 160, 2035–2043 (2002).
- Kaab S, Barth AS, Margerie D et al. Global gene expression in human myocardium–oligonucleotide microarray analysis of regional diversity and transcriptional regulation in heart failure. J. Mol. Med. 82, 308–316 (2004).