

Fetal glucocorticoid overexposure (exogenous administration of glucocorticoids or inhibition of feto-placental 11b-HSD2 activity), in combination with genetic factors, will influence the development of many organ systems, and program tissue responses in the developing offspring, leading to adult disorders.
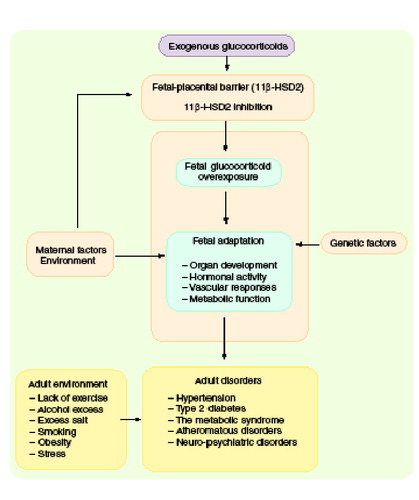


Traditionally, lifestyle and genetic predisposition are considered to be the main determinants of the common non communicable cardiovascular and metabolic diseases of middle age, such as hypertension, ischemic heart disease and Type 2 diabetes mellitus. However, over the last decade an increasing number of epidemiological studies have suggested that factors operating in early-life are also important determinants of the risk of cardiovascular and metabolic disorders in adulthood. This organizational phenomenon is known as early-life programming. Data from several distinct populations in Europe, Asia, Australia and North America have shown that low birth-weight strongly predicts the subsequent occurrence of hypertension, hyperlipidemia, insulin resistance, Type 2 diabetes and ischemic heart disease deaths in adult life Citation[1]. These relationships are largely independent of traditional adult lifestyle factors (e.g., smoking, adult weight, social class, excess alcohol intake and sedentariness), which are additive risks to the effects of birth weight.
Early-life programming
The concept of developmental physiological programming or imprinting has been advanced to explain the associations between prenatal environmental events, altered fetal growth and development and later pathophysiology Citation[2,3]. Programming reflects the action of a factor during a sensitive developmental period or window to affect the development and organization of specific tissues that are concurrently vulnerable, producing effects that persist throughout life. Different cells and tissues are sensitive at different times, so the effects of environmental challenges will be distinct, depending on both the challenge involved and also its timing.
Programming has been examined in several experimental mammalian systems. Many agents including genes, transcription factors, growth factors, hormones, stress and nutrients are known to exert programming actions. These effects are often found in combination with alterations in steroidal hormones, and one of the best characterized programming effects involves the action of androgens. Neonatal exposure to androgens programs the development of sexually dimorphic structures in the brain and facilitates male-type sexual behavior. These effects persist throughout life, irrespective of subsequent hormonal manipulations or the genetic sex of the animal.
The molecular mechanisms for the association between low birth-weight and later disease are unknown, but two major environmental hypotheses have been proposed to underlie mammalian fetal programming: fetal malnutrition Citation[2] and overexposure of the fetus to glucocorticoids Citation[3].
Glucocorticoids & fetal development
Glucocorticoids (cortisol in man; corticosterone in rats) are lipophilic molecules produced by the adrenal cortex and exert their effects by binding to intracellular glucocorticoid receptors (GRs), which act in the cell nucleus to regulate the expression of myriad target genes. In adult mammals, glucocorticoids are involved in the control of several physiological processes that maintain homeostasis, including coordination of responses to stress. Excessive glucocorticoids, either from endogenous overproduction in Cushing’s syndrome or as a result of exogenous administration, have well-characterized diabetogenic and hypertensive effects, as summarized in .
During development, glucocorticoids are important for growth, tissue development and maturation of various organs to prepare the organism for an extra-uterine life. The ability of glucocorticoids to accelerate maturation of organs, notably the lung, accounts for their widespread use in obstetric and neonatal practice in threatened or actual preterm delivery to improve neonatal viability. However, supraphysiological levels of glucocorticoids cause fetal growth retardation in mammalian models and humans. Human intrauterine growth retardation is associated with high maternal and fetal concentrations of glucocorticoids. Moreover, in utero levels of glucocorticoids are also increased in response to most challenges known to have programming effects, such as maternal undernutrition, placental insufficiency and restriction of placental blood flow. Glucocorticoids may represent one of a limited range of signaling options between mother, placenta and fetus, aiding adaptation of placental function and fetal development to maximize the chances of survival at birth and, thereafter, infant growth, behavior and reproductive strategy. Indeed, disease in the post reproductive years may merely be a remote echo of the predictive adaptation that is hypothesized to underpin the occurrence of developmental programming in many species from simple invertebrates to mammals Citation[4].
Feto–placental 11β-hydroxysteroid dehydrogenase Type 2: a physiologic barrier
Fetal glucocorticoid levels are normally much lower than maternal levels Citation[5]. This gradient is achieved by feto–placental 11β-hydroxysteroid dehydrogenase Type 2 (11β-HSD2), which catalyzes the conversion of cortisol and corticosterone to physiologically inert 11-keto forms: cortisone and 11-dehydrocorticosterone Citation[6].
In the placenta, 11β-HSD2 forms a potent barrier to maternal glucocorticoids, although the barrier may not be complete as a minor proportion of maternal glucocorticoid crosses intact to the fetus Citation[6]. The mechanisms involved in the regulation of placental 11β-HSD2 are largely unknown. A deficiency in 11β-HSD2 would be expected to expose the fetus to increased glucocorticoid levels from the maternal circulation, with subsequent effects on fetal development Citation[3]. In support of this notion, the lowest placental 11β-HSD2 activity, and presumably the highest fetal exposure to maternal glucocorticoids, is seen in human babies with the smallest birth weights Citation[7], mirroring similar observations in rodents Citation[8]. Moreover, patients bearing mutations of the gene encoding 11β-HSD2 have low birth weight. Fetal glucocorticoid load can be artificially increased by maternal administration of a synthetic glucocorticoid, such as dexamethasone (a poor substrate for 11β-HSD2) or through inhibition of placental 11β-HSD2 by liqorice and its derivatives (e.g., carbenoxolone) Citation[9]. Interestingly, maternal protein restriction also reduces the activity of 11β-HSD2 in the placenta Citation[10], which presumably results in increased fetal glucocorticoid load. This effect on an important placental enzyme provides further support that glucocorticoids may play a role in the effects of maternal undernutrition in programming later pathophysiology.
Given the evidence outlined above for developmental actions of prenatal glucocorticoid exposure, might these associate with persisting effects in the offspring?
Programming cardiovascular & metabolic systems
Blood pressure, kidney & heart
The impact of prenatal glucocorticoid exposure during development on arterial blood pressure has been investigated in the offspring of several species. Treatment of pregnant rats with dexamethasone results in persistent elevations of arterial blood pressure in the adult offspring Citation[8]. Maternal administration of carbenoxolone, a potent inhibitor of 11β-HSD, also leads to elevated blood pressure in the adult offspring Citation[9]. This effect requires the presence of maternal glucocorticoids, as the offspring of adrenalectomized pregnant rats are protected from carbenoxolone actions upon adult hypertension.
The timing of glucocorticoid exposure appears to be important; exposure to glucocorticoids during the final week of pregnancy in the rat is sufficient to produce permanent adult hypertension, whereas the sensitive window for such effects in sheep is earlier in gestation Citation[11]. The reasons for such differences are unclear but may reflect the complex species-specific patterns of expression of glucocorticoid receptors and the isozymes of 11β-HSD2, which regulate maternal glucocorticoid transfer to the fetus and modulate glucocorticoid action in individual developing tissues.
The mechanisms of glucocorticoid-programmed adult hypertension are not clearly understood, but probably involve a variety of processes. Potential mechanisms may involve changes in renal structure (irreversible reductions in nephron number), changes in vascular responsivity, enhanced activity of the renin–angiotensin system, alteration of the baroreceptor response and primary effects on the heart by interfering with the development of cardiac noradrenergic innervation and sympathetic activity (reviewed Citation[12]).
Glucose–insulin homeostasis & metabolism
Prenatal overexposure to glucocorticoids also causes permanent hyperglycemia and hyperinsulinemia in the adult offspring in the rat and sheep Citation[13,14]. Like blood pressure, the window of sensitivity for programming hyperglycemia in the rat occurs in the final third of gestation. Earlier dexamethasone exposure or postpartum treatments do not program hyperglycemia/hyperinsulinemia, suggesting that there is a tight window for this effect, though again species differ in the timing of sensitivity. The mechanisms through which prenatal exposure to excess glucocorticoids programs hyperglycemia have not been fully determined, but may involve permanent alterations in gene expression in key metabolic target organs such as the liver, pancreas, fat and skeletal muscle. For instance, glucocorticoids regulate expression of critical hepatic metabolic enzymes, notably phosphoenolpyruvate carboxykinase (PEPCK), which develops in late gestation and catalyzes a rate-limiting step in gluconeogenesis and whose activity is increased in patients with Type 2 diabetes. In rats, exposure to excess dexamethasone in utero leads to offspring with permanent elevations in PEPCK mRNA and enzyme activity Citation[13]. Interestingly, the effects of prenatal dexamethasone treatment are not only observed in the immediate offspring as adults, but the second-generation offspring also show elevated PEPCK and insulin levels, without further dexamethasone exposure Citation[15]. The mechanisms for this transgenerational transmission of the phenotype are unknown, but may involve epigenetic processes.
Relatively little is known about the effects of prenatal exposure to glucocorticoids on the development of the endocrine functions of the pancreas. However, indirect evidence suggests that excessive prenatal glucocorticoid levels may cause long-term β-cell dysfunction Citation[16]. Prenatal dexamethasone exposure in rats programs fat metabolism. Adult offspring of dams exposed to dexamethasone in pregnancy have increased intra-abdominal fat depots, with a parallel increase in leptin levels. The same antenatal treatment may also affect skeletal muscle glucose metabolism.
Programming of the CNS, the hypothalamic–pituitary–adrenal axis & behavior
Glucocorticoids are important for normal development in most regions of the CNS, initiating terminal maturation and remodeling axons and dendrites, and for cell survival. Prenatal glucocorticoid administration retards brain weight at birth in sheep, delaying maturation of neurons, myelination, glia and vasculature Citation[17].
Exposure to glucocorticoids in utero has widespread acute effects upon neuronal structure and synapse formation, and may permanently alter brain structure. Fetal rhesus monkeys administered dexamethasone exhibited marked atrophy of hippocampal cells at birth, and their hippocampi failed to recover to normal size even at 2 years postpartum. At birth, the brain of the neonatal rhesus monkey is 60% of adult size, whereas the human brain is only 24% of adult size. Thus, the impact of prenatal dexamethasone overexposure on the developing nervous system may be of a greater magnitude in the young monkey.
The hypothalamo–pituitary circuit, which is central to the integration of the individual’s endocrine and behavioral responses, as well as various functions critical for the body’s homeostasis, appears highly sensitive to excess glucocorticoid exposure during development. A number of animal studies have shown that exposure to synthetic glucocorticoids in utero results in both disturbed hypothalamo–pituitary functioning and long-term alterations in the set points of several major hormonal axes, including adrenal glucocorticoid secretion Citation[18].
As revealed, mainly by studies in rats, overexposure to glucocorticoids in utero leads to alterations in adult behavior. Late gestational dexamethasone or 11β-HSD inhibitors in rats impairs coping in aversive situations later in life Citation[19,20]. Behavioral changes in adults exposed prenatally to glucocorticoids may be associated with altered functioning of the amygdala, a key structure in the regulation of fear and anxiety. Interestingly, adult offspring of dexamethasone-treated dams show increased corticotropin-releasing hormone levels, specifically in the central nucleus of the amygdala.
The glucocorticoid receptor gene: a common programming target
The GR is critical for cell function and GR gene expression shows marked tissue-specific regulation. In the peripheral tissues, such as the liver and visceral adipose tissue, prenatal glucocorticoid exposure causes a permanent increase in GR expression and this upregulation is paralleled by increased glucocorticoid sensitivity in these tissues Citation[13].
A critical question that remains largely unanswered is how discrete perinatal environmental events such as glucocorticoid exposure can permanently alter gene expression. Recent evidence suggests that it may involve selective methylation/demethylation of specific promoters of the GR gene. Changes in the GR promoter DNA methylation pattern were associated with altered histone acetylation and transcription factor (NGFI-A) binding to the GR promoter. Treatment of the adult rat offspring with a histone deacetylase inhibitor removes the epigenetic differences in histone acetylation and DNA methylation and hence the NGFI-A binding changes Citation[21]. During development, such target promoter demethylation occurs before birth and may fine-tune the promoter to memorize regulatory events occurring during development. This novel mechanism of gene control by environmental events early in life, which then persists throughout the lifespan, remains to be confirmed in other species.
Evidence for glucocorticoid programming in humans
Glucocorticoids such as dexamethasone and betamethasone have extensive therapeutic use in obstetric practice; they are used as immunosuppressants to control various maternal conditions such as connective tissue disorders, as well as for short-term treatment of the fetus to accelerate lung maturation in cases of preterm labor to prevent neonatal respiratory distress syndrome. More rarely, dexamethasone is used throughout gestation from the first trimester to attenuate fetal adrenal steroid overproduction in fetuses at risk of congenital adrenal hyperplasia. The long-term effects of fetal glucocorticoid exposure in humans have still not been comprehensively investigated. A preliminary study in children at risk from congenital adrenal hyperplasia, who received dexamethasone in early gestation showed that prenatal dexamethasone exposure did not significantly affect cognition, but these children had increased emotional and social behavior problems Citation[22]. The effects of fetal glucocorticoid exposure on cardiovascular and metabolic function in humans are rarely reported, but what data exist suggest some elevation of blood pressure Citation[23] and insulin levels Citation[24] many years after even brief prenatal therapy. Proper evaluation of long-term effects of prenatal glucocorticoid exposure in humans awaits rigorous follow-up studies.
Therapeutic thoughts
Recently, a number of investigators have begun to explore methods to reverse prenatal programming by environmental insults. Leptin appears to obviate adverse metabolic outcomes Citation[25], and maternofetal manipulations may exert similar effects on neuroendocrine parameters. Intriguingly, the insulin-sensitizing drug metformin may specifically normalize programmed changes in GR expression in the liver Citation[26]. Whether or not any of these are potentially useful in humans remains uncertain.
Conclusions
The main points of this article are summarized in . Prenatal exposure to glucocorticoids may program a range of tissue-specific pathophysiologies, with cardiometabolic and CNS effects predominating. The finetuning of fetal physiology by the environment is conserved and therefore important. Studies to unravel the underlying processes are a prerequisite for rational treatments for the consequences of adverse developmental environments.
Acknowledgements
The authors’ research is in part supported by a grant from the European Commission (QLRT-2001–02758; EUPEAH). See also www.eupeah.org.
References
- Barker DJP. The developmental origins of well-being. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 359 (1449), 1359–1366 (2004).
- Barker DJP, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet 341, 938–941 (1993).
- Edwards CRW, Benediktsson R, Lindsay R, Seckl JR. Dysfunction of the placental glucocorticoid barrier: a link between the foetal environment and adult hypertension? Lancet 341, 355–357 (1993).
- Gluckman P, Hanson M. Living with the past: evolution, development, and patterns of disease. Science 305, 1733–1736 (2004).
- Beitens IZ, Bayard F, Ances IG, Kowarski A, Migeon CJ. The metabolic clearance rate, blood production, interconversion and transplacental passage of cortisol and cortisone in pregnancy near term. Pediatr. Res. 7, 509–519 (1973).
- Benediktsson R, Calder AA, Edwards CRW, Seckl JR. Placental 11β-hydroxysteroid dehydrogenase Type 2 is the placental barrier to maternal glucocorticoids: ex vivo studies. Clin. Endocrinol. 46, 161–166 (1997).
- Stewart PM, Rogerson FM, Mason JI. Type 2 11β-hydroxysteroid dehydrogenase messenger RNA and activity in human placenta and fetal membranes: its relationship to birth weight and putative role in fetal steroidogenesis. J. Clin. Endocrinol. Metab. 80, 885–890 (1995).
- Benediktsson R, Lindsay R, Noble J, Seckl JR, Edwards CRW. Glucocorticoid exposure in utero: a new model for adult hypertension. Lancet 341, 339–341 (1993).
- Lindsay RS, Lindsay RM, Edwards CRW, Seckl JR. Inhibition of 11β-hydroxysteroid dehydrogenase in pregnant rats and the programming of blood pressure in the offspring. Hypertension 27, 1200–1204 (1996).
- Langley-Evans SC, Philips G, Benediktsson R et al. Maternal dietary protein restriction, placental glucocorticoid metabolism and the programming of hypertension. Placenta 17, 169–172 (1996).
- Dodic M, May CN, Wintour EM, Coghlan JP. An early prenatal exposure to excess glucocorticoid leads to hypertensive offspring in sheep. Clin. Sci. 94, 149–155 (1998).
- Seckl J. Prenatal glucocorticoids and long-term programming. Eur. J. Endocrinol. 151, U49–U62 (2004).
- Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR. Glucocorticoid exposure in late gestation permanently programmes rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J. Clin. Invest. 101, 2174–2181 (1998).
- Moss TJ, Sloboda DM, Gurrin LC, Harding R, Challis JR, Newnham JP. Programming effects in sheep of prenatal growth restriction and glucocorticoid exposure. Am. J. Physiol.281(3), R960–R970 (2001).
- Drake AJ, Walker BR, Seckl JR. Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am. J. Physiol. 288, R34–R38 (2005).
- Blondeau B, Lesage J, Czernichow P, Dupouy JP, Breant B. Glucocorticoids impair fetal β-cell development in rats. Am. J. Physiol. 281(3), E592–E599 (2001).
- Huang WL, Beazley LD, Quinlivan JA, Evans SF, Newnham JP, Dunlop SA. Effect of corticosteroids on brain growth in fetal sheep. Obstet. Gynecol. 94(2), 213–218 (1999).
- Matthews SG. Early programming of the hypothalamo-pituitary-adrenal axis. Trends Endocrinol. Metab. 130, 373–380 (2002).
- Welberg LAM, Seckl JR, Holmes MC. Inhibition of 11β-hydroxysteroid dehydrogenase, the feto-placental barrier to maternal glucocorticoids, permanently programs amygdala glucocorticoid receptor mRNA expression and anxiety-like behavior in the offspring. Eur. J. Neurosci. 12, 1047–1054 (2000).
- Welberg LAM, Seckl JR, Holmes MC. Prenatal glucocorticoid programming of brain corticosteroid receptors and corticotrophin-releasing hormone: possible implications for behavior. Neuroscience 104, 71–79 (2001).
- Weaver I, Cervoni N, Champagne F et al. Epigenetic programming by maternal behavior. Nature Neurosci. 7, 847–854 (2004).
- Trautman PD, Meyer-Bahlburg HFL, Postelnek J, New MI. Effects of early prenatal dexamethasone on the cognitive and behavioral development of young children: results of a pilot study. Psychoneuroendocrinology 20, 439–449 (1995).
- Doyle LW, Ford GW, Davis NM, Callanan C. Antenatal corticosteroid therapy and blood pressure at 14 years of age in preterm children. Clin. Sci. 98, 137–142 (2000).
- Dalziel SR, Walker NK, Parag V. Cardiovascular risk factors after antenatal exposure to betamethasone: 30-year follow-up of a randomised controlled trial. Lancet 365(9474), 1856–1862 (2005).
- Vickers MH, Gluckman PD, Coveny AH et al. Neonatal leptin treatment reverses developmental programming. Endocrinology 146(10), 4211–4216 (2005).
- Cleasby ME, Livingstone DEW, Nyirenda MJ, Seckl JR, Walker BR. Is programming of glucocorticoid receptor expression by prenatal dexamethasone in the rat secondary to metabolic derangement in adulthood? Eur. J. Endocrinol.. 148(1), 129–138 (2003).