
Abstract
Common variable immunodeficiency (CVID) is a heterogeneous group of disorders, characterized by decreased serum levels of immunoglobulins and abnormal antibody response to protein and/or polysaccharide antigens, leading to recurrent respiratory and gastrointestinal infections, autoimmunity and malignancies. Meanwhile, several monogenic defects with CVID-like phenotype have been identified during the last decade. There is a need to reach international consensus by modifying criteria for CVID, considering various areas of uncertainty in the field.
Common variable immunodeficiency (CVID), the most common life-threatening primary immunodeficiency disease (PIDD), was described six decades ago Citation[1]. It is defined as low immunoglobulin levels and abnormal antibody response to protein and/or polysaccharide antigens Citation[2–4]. CVID patients suffer from recurrent infections, autoimmunity, lymphoproliferation and malignancies Citation[3,5–7]. The underlying gene defect(s) are mostly unknown Citation[8]. However, 60 years after the original report significant progress in treating these patients has resulted in markedly better prognosis and quality of life Citation[7,9].
CVID is a heterogeneous group of presumably mostly polygenic diseases Citation[10]. Monogenic defects that cause (ICOS, CD19, CD20, CD81) or predispose to (TACI, BAFFR, MSH5) CVID phenotype, or cause combined PIDD that may at first resemble CVID (LRBA, PRKCD, TWEAK, PI3K, STAT1 GOF), have been increasingly described. Also, the numbers of agammaglobulinemia (BTK, IGHM, IGLL1, CD79A, CD79B, PI3K, BLNK, TCF3) and class switch recombination deficiency genes (CD40L, CD40, AID, UNG, PI3K) have increased; some genes are able to cause various phenotypes . For many of these monogenic diseases, the exact clinical phenotype is not yet known due to the small number of patients described. However, identification of such monogenic diseases suggests revisiting the patients previously known as CVID.
Figure 1. Approach to a patient with hypogammaglobulinemia.
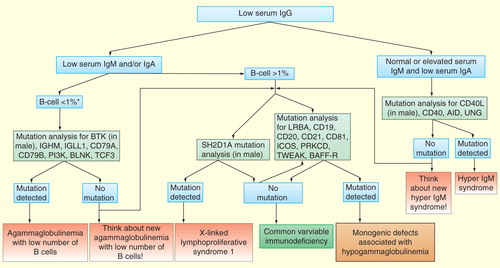
Many units do not routinely perform genetic testing in patients with CVID phenotype. This leads to unsatisfactory knowledge about different genetic forms, their true prevalence and geographic distribution. Genetic testing may be deemed unnecessary or cost-prohibitive, though testing potentially leads to increased knowledge, improved genetic counseling, targeted follow-up and therapy. In our opinion, the reluctance to perform genetic testing, especially in children, can lead to delayed treatment and potentially deleterious mistakes, if monogenic combined PIDD are not recognized early. Various other PIDD, including severe combined and combined immunodeficiencies, X-linked lymphoproliferative syndromes, autoimmune lymphoproliferative syndromes, familial hemophagocytic lymphohistiocytosis syndromes and autoinflammatory syndromes (hyper IgD syndrome, PLCG2-associated antibody deficiency and immune dysregulation) may initially be mislabeled as CVID.
Since most PIDDs are highly heterogeneous, with the same gene causing different phenotypes even within the family and different genes causing similar phenotypes, the constraints of setting a definite diagnosis purely on clinical grounds are considerable. With the decreasing cost of genome-wide exome sequencing, this problem may soon become obsolete. In the authors’ opinion, known monogenic diseases should be named according to their causative genes, functional mechanisms (e.g., gain of function, hypomorphic), patterns of inheritance (e.g., autosomal dominant, dominant negative) and phenotype; they should not be labeled as CVID anymore. Essentially, many CVID diagnoses will remain those of exclusion, based on a matching phenotype, decrease of immunoglobulin levels and vaccine responses. Low number of B cells in a subset of reported CVID patients is a challenging issue. It is unclear whether these patients represent true CVID or a form of agammaglobulinemia with low number of B cells.
Transient hypogammaglobulinemia of infancy (THI) is defined as hypogammaglobulinemia due to a delay in infant’s own IgG production after the disappearance of maternal transplacental IgG Citation[4]. It was believed that this could extend only into the second year of life; hypogammaglobulinemia lasting longer would be CVID. According to recent reports, THI may take up to 4 years to resolve and only this long follow-up will eventually differentiate between THI and CVID. Although asymptomatic infants with THI do not require any treatment, IgG replacement therapy (IgGRT) is needed for patients with hypogammaglobulinemia and recurrent and severe infections, at any ages.
Closer study of the known monogenic primary antibody deficiencies (PAD) reveals that these too are highly heterogeneous. A significant proportion of patients do not have CVID-like phenotype, but have – according to laboratory results – milder forms of PAD. Despite this, they still may have severe complications from disease. According to the Pan-American Group for Immunodeficiency and European Society for Immunodeficiencies criteria, published more than a decade ago, these patients may be labeled as ‘possible CVID’, as ‘specific antibody deficiency’ or they have something ‘in between’ Citation[2]. For a considerable proportion of patients admitted for evaluation, we have no generally accepted terms. These patients have been labeled as ‘non-specific’ or idiopathic hypogammaglobulinemia, graded according to locally defined prioritizing protocols for severity, or given no descriptive terms Citation[11–14]. In the authors’ opinion, these patients should not be named CVID. Any CVID case should have clinical signs, symptoms and matching laboratory findings to establish the diagnosis. This becomes especially important when conducting scientific studies, to avoid excessive heterogeneity and potential dilution of therapeutic effect if milder cases are mislabeled.
The diagnosis of any PAD heavily depends on the best presently available functional tests (anti-tetanus, anti-diphtheria and anti-pneumococcal polysaccharide responses), with a number of unavoidable caveats to interpretation of anti-pneumococcal polysaccharide (PnP) responses Citation[15]. A number of patients with PAD could have normal antibody responses to PnP, possibly with a defect in antibody-mediated opsonophagocytosis not shown by routine tests Citation[16,17]. In clinical practice, patients with very low immunoglobulin levels and a full CVID phenotype may still have a defect in solely T-independent anti-PnP or only anti-PnP and anti-diphtheria may be low. Whether solely low anti-PnP responses, low immunoglobulin levels and CVID phenotype justify the diagnosis of CVID is not a matter of general consensus. In the authors’ experience, response to IgG therapy does not seem to differ from those with low T-dependent responses as well.
The most painful disagreement in diagnosing PAD patients is exactly how low anti-PnP responses should be. While the current American Academy of Allergy Asthma and Immunology (AAAAI) criteria define non-protective anti-PnP titers using level IIb–III evidence and a threshold of >1.3 μg/ml, there are no large systematic trials to substantiate these criteria Citation[18]. An ongoing British-Finnish trial has systematically looked into this; the preliminary results were first presented in European Society for Immunodeficiencies 2010 in Istanbul. The results suggest a threshold of >0.34 μg/ml; this is backed up by multiple vaccine studies as protective against invasive disease (Kumararatne D, PERS. COMM.). We most likely have a large patient group, some of whom may be receiving IgGRT, with borderline low (defined as falling in between these two criteria) vaccine responses and low immunoglobulin levels. We have no good follow-up data in such patients to suggest therapeutic effect or prolonged life expectancy. Like in literature, the authors have found borderline vaccine responses in patients without CVID phenotype, sometimes with asthma, recurrent upper respiratory infections, chronic fatigue and/or long-standing depression as clinical complaints Citation[15]. Whether a subset of such patients with for example recurrent pneumonias and/or bronchiectasis would benefit clearly from IgG replacement has not been studied formally.
In science and medicine, one often needs to start descriptively, to properly assess the prevalence and severity of a problem. Highly likely any immunodeficiency unit having success in increasing the level of public awareness on PIDD in its area will meet many such borderline patients. Some of them may even have a known monogenic defect Citation[11–14]. During follow-up, some do develop a full-blown CVID phenotype and the delay in treatment becomes minimal. Some have end-organ complications (like invasive infections and bronchiectasis) already at presentation, or they may remain stable with antibiotic prophylaxis. After six decades of studies on PAD, we still have no terms to describe these patients. In our opinion, this is not good clinical practice but bad science.
Outside probable CVID, agammaglobulinemias and hyper-IgM syndromes, we inadequately know when to treat. Acknowledgement of this has lead to implementation of local grading systems to target the most needy with this invaluable resource Citation[14]. If treatment is considered, we probably should study and target patients with clear lung damage first and acknowledge our present incapability to help many patients with milder forms of PAD and chronic rhinosinusitis with purulent exacerbations. However, to tackle this conundrum properly, we should first agree on how to describe these PAD patients phenotypically, then conduct proper treatment trials and search for prognostic factors to aid clinical decisions (e.g., B-cell subsets, end-organ damage, nature of infections). In the process, we should not assume that with any PAD diagnosis automatically comes IgGRT. New criteria to define CVID have been recently proposed Citation[12]. To consolidate different criteria and views to interpret vaccine responses, we may need to define a definite CVID patient as someone without known monogenic defects, with severely low vaccine responses and immunoglobulin levels together with classical phenotype. Similar findings but borderline responses would justify a diagnosis of possible CVID.
There is enormous geographic variation in when to treat and whom, and the authors believe this to be partly caused by our classification schemes and terminology failing to meet clinical and study demands. For a busy clinician, it may be just that much easier to simply treat. Though IgG is life saving in many diseases, an increasing demand and limited supply has resulted in soaring prices Citation[14]. Increasing demand and prices may leave CVID patients in less-affluent countries without therapy.
CVID manifesting after Evans syndrome has traditionally been regarded as primary. With the increasing use of B-cell antibodies in the treatment of hematologic autoimmune diseases and lymphomas, there seems to be an increasing number of persistent CVID-like phenotypes remitted for evaluation. This impression is partly supported by recent studies Citation[19,20]. Hypogammaglobulinemia may disappear up to 18 months after cessation of rituximab therapy Citation[21]. In authors’ opinion, a persistent CVID-like phenotype after 18 months of therapy in these patients warrants therapy with IgGRT but more studies, longer follow-up and more insight into potential genetic susceptibility to PAD in these patients is needed.
Differentiation of primary and secondary cases becomes ever more difficult, if milder forms of PAD are assessed, especially isolated low IgG. Though CVID patients often have obstructive lung disease, secondary hypogammaglobulinemia caused by long-term corticosteroid use in steroid-dependent asthma can be tough to diagnose Citation[22]. There seems to be insufficient data on differences between glucocorticoid preparations and inter-individual variation resulting in low IgG and/or missing polysaccharide responses during oral and inhaled steroid use in a significant proportion of asthmatics, described to last for up to 2 years Citation[23]. In clinical practice, corticosteroids often seem to lower anti-PnP responses to those reaching AAAAI criteria, but this has not been formally studied. Intravenous immunoglobulin use in corticosteroid-dependent asthma results in serious side effects Citation[24]. Also, the failure to exclude vitamin B12 deficiency, vitamin A deficiency or hypothyroidism in patients as a cause of low IgG and missing polysaccharide responses has lead to severe angioedema and worsening asthma with IgGRT, ameliorated only by cessation of therapy [Seppänen M, Aghamohammadi I, Rezaei N, Unpublished Data]. Potentially, immunomodulation caused by high-dose IgGRT to asthmatics satisfying AAAAI criteria may deleteriously intensify Th2 responses Citation[25,26]. Before diagnosis of primary PAD, if any IgGRT is considered, one needs to evaluate the patient carefully; treatment of steroid-induced low IgG, in authors’ opinion, is rarely warranted Citation[27].
Therefore in authors’ opinion, definite CVID should be considered in any patient older than (2-) 4 years with significantly decreased level of IgG (at least 2 standard deviation below the mean for age) in addition to decrease in IgM and/or IgA, clearly poor antibody responses to vaccines and/or isohemagglutinin level(s), after exclusion of other causes of hypogammaglobulinemia (Box 1). We should, however, strive to reach international consensus by modifying criteria for PAD. Various areas of uncertainty should be acknowledged in the process. Patients with borderline vaccine responses or solely low IgG should be included into the nomenclature; this will enable proper scientific studies on etiology and therapy.
Box 1. Proposed modified criteria for common variable immunodeficiency.
Decreased serum IgG level (at least 2 standard deviation below the mean for age)
Decreased serum IgM and/or IgA levelFootnote †
Decreased response to vaccines and/or absent isohemagglutinins level(s)
B cell number of >1% of total lymphocytesFootnote ‡
Age of 4 years or more at the time of diagnosis
Exclusion of other causes of hypogammaglobulinemia
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending or royalties.
No writing assistance was utilized in the production of this manuscript.
Notes
†Patients with normal or low B cells in blood, decreased IgG (and possibly missing IgE), but normal IgA and IgM levels in serum, clearly deficient responses to vaccines and without monogenic defect or discernible secondary causes should be defined as suffering from 'primary IgG hypogammaglobulinemia'. During follow-up, some of these patients may further develop common variable immunodeficiency.
‡Patients with very low B cells (<1%) should be investigated for the genes associated with agammaglobulinemia and low number of B cells.
Related Research Data
References
- Janeway CA, Apt L, Gitlin D. Agammaglobulinemia. Trans. Assoc. Am. Physicians 66, 200–202 (1953).
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin. Immunol. 93(3), 190–197 (1999).
- Aghamohammadi A, Parvaneh N, Rezaei N. Common variable immunodeficiency: a heterogeneous group needs further subclassification. Expert Rev. Clin. Immunol. 5(6), 629–631 (2009).
- Aghamohammadi A, Lougaris V, Plebani A, Miyawaki T, Durandy A, Hammarström L. Predominantly antibody deficiencies. In: Primary Immunodeficiency Diseases: Definition, Diagnosis and Management. Rezaei N, Aghamohammadi A, Notarangelo LD (Eds). Springer-Verlag Berlin, Heidelberg, Germany, 97–130 (2008).
- Aghamohammadi A, Abolhassani H, Moazzami K, Parvaneh N, Rezaei N. Correlation between common variable immunodeficiency clinical phenotypes and parental consanguinity in children and adults. J. Investig. Allergol. Clin. Immunol. 20(5), 372–379 (2010).
- Aghamohammadi A, Farhoudi A, Moin M et al. Clinical and immunological features of 65 Iranian patients with common variable immunodeficiency. Clin. Diagn. Lab. Immunol. 12(7), 825–832 (2005).
- Chapel H, Lucas M, Lee M et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 112(2), 277–286 (2008).
- Al-Herz W, Bousfiha A, Casanova JL et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front. Immunol. 2, 54 (2011).
- Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin. Immunol. 137(1), 21–30 (2010).
- Orange JS, Glessner JT, Resnick E et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J. Allergy Clin. Immunol. 127(6), 1360–1367.e1366 (2011).
- Agarwal S, Cunningham-Rundles C. Treatment of hypogammaglobulinemia in adults: a scoring system to guide decisions on immunoglobulin replacement. J. Allergy Clin. Immunol. 131(6), 1699–1701 (2013).
- Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous Immunoglobulin. Clin. Exp. Immunol. 174(2), 203–211 (2013).
- Driessen GJ, Dalm VA, van Hagen PM et al. Common variable immunodeficiency and idiopathic primary hypogammaglobulinemia: two different conditions within the same disease spectrum. Haematologica 98(10), 1617–123 (2013).
- Orange JS, Ochs HD, Cunningham-Rundles C. Prioritization of evidence-based indications for intravenous immunoglobulin. J. Clin. Immunol. 33(6), 1033–1036 (2013).
- Gelfand EW, Ochs HD, Shearer WT. Controversies in IgG replacement therapy in patients with antibody deficiency diseases. J. Allergy Clin. Immunol. 131(4), 1001–1005 (2013).
- Rezaei N, Aghamohammadi A, Siadat SD et al. Serum bactericidal antibody responses to meningococcal polysaccharide vaccination as a basis for clinical classification of common variable immunodeficiency. Clin. Vaccine Immunol. 15(4), 607–611 (2008).
- Rezaei N, Siadat SD, Aghamohammadi A et al. Serum bactericidal antibody response 1 year after meningococcal polysaccharide vaccination of patients with common variable immunodeficiency. Clin. Vaccine Immunol. 17(4), 524–528 (2010).
- Orange JS, Ballow M, Stiehm ER et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the basic and clinical immunology interest section of the American Academy of Allergy, Asthma & Immunology. J. Allergy Clin. Immunol. 130(3 Suppl.), S1–S24 (2012).
- Casulo C, Maragulia J, Zelenetz AD. Incidence of hypogammaglobulinemia in patients receiving rituximab and the use of intravenous immunoglobulin for recurrent infections. Clin. Lymphoma Myeloma Leuk. 13(2), 106–111 (2013).
- Kano G, Nakatani T, Yagi K, Sakamoto I, Imamura T. Complicated pathophysiology behind rituximab-induced persistent hypogammaglobulinemia. Immunol. Lett. doi:10.1016/j.imlet.2013.10.005 (2013) ( Epub ahead of print).
- Torgerson TR, Bonagura VR, Shapiro RS. Clinical ambiguities--ongoing questions. J. Clin. Immunol. (33 Suppl. 2), S99–S103 (2013).
- Urm SH, Yun HD, Fenta YA et al. Asthma and risk of selective IgA deficiency or common variable immunodeficiency: a population-based case-control study. Mayo Clin. Proc. 88(8), 813–821 (2013).
- Kawano T, Matsuse H, Obase Y et al. Hypogammaglobulinemia in steroid-dependent asthmatics correlates with the daily dose of oral prednisolone. Int. Arch. Allergy Immunol. 128(3), 240–243 (2002).
- Kishiyama JL, Valacer D, Cunningham-Rundles C et al. A multicenter, randomized, double-blind, placebo-controlled trial of high-dose intravenous immunoglobulin for oral corticosteroid-dependent asthma. Clin. Immunol. 91(2), 126–133 (1999).
- Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature 475(7354), 110–113 (2011).
- Grotenboer NS, Ketelaar ME, Koppelman GH, Nawijn MC. Decoding asthma: translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J. Allergy Clin. Immunol. 131(3), 856–865 (2013).
- Cunningham-Rundles C. Key aspects for successful immunoglobulin therapy of primary immunodeficiencies. Clin. Exp. Immunol. 164 (Suppl. 2), 16–19 (2011).