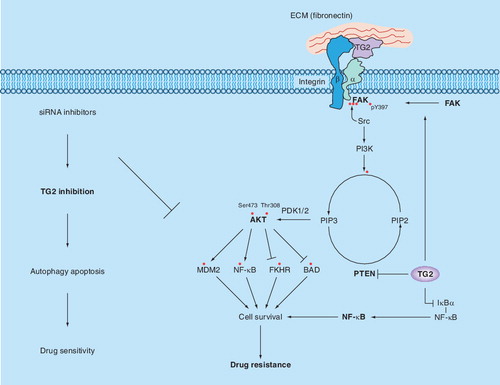
Association of TG2 with integrins (e.g., β1, β3, β4 or β5) can increase their avidity for extracellular matrix proteins, such as fibronectin, and result in the activation of downstream cell survival FAK/Akt signaling pathways. TG2 can also affect FAK phosphorylation directly by associating with FAK or indirectly by promoting PTEN degradation via the ubiquitin–proteasomal pathway. Either collectively or individually, activation of these signaling molecules in the FAK/Akt axis leads to the phosphorylation of various downstream substrates and activation of MDM2 and NF-κB or inhibition of FKHR and BAD proteins. Collectively, these events contribute to increased cell survival and chemoresistance. Conversely, TG2 – by crosslinking IκBα (inhibitor of κBα) – can induce dissociation of the NF-κB–IκBα complex, resulting in constitutive activation of NF-κB in an IKK-independent manner.
FAK: Focal adhesion kinase; TG2: Tissue transglutaminase.
Ovarian cancer remains the most common cause of gynecologic malignancy-related death in the USA Citation[1]. The poor outcomes associated with ovarian cancer are due to the frequent presence of advanced-stage disease and widespread peritoneal metastasis at the time of diagnosis. While most ovarian cancer patients respond to initial treatment with cytoreductive surgery and systemic chemotherapy, more than 70% experience a recurrence and eventually die of complications resulting from drug failure Citation[2]. Therefore, to improve available treatments, develop new therapeutic options and improve patients’ outcomes, it is important to understand the biological mechanisms that underlie the recurrence of ovarian cancer and development of chemotherapy resistance.
A current working model for metastases from ovarian cancer suggests that cancer cells exfoliate from the ovaries into the peritoneum fluid, where they adhere to mesothelial cells lining the inner surface of the peritoneal cavity. Once the cancer cells attach to the mesothelial cells, they migrate through the layers of mesothelial cells, invade the local organs and spread to distant sites. The process of cancer cell adhesion, migration and invasion is accompanied by several alterations at the interface between tumor cells and the peritoneum. These alterations promote dynamic interactions among cell adhesion molecules, the extracellular matrix (ECM), cytoskeletal proteins and signaling molecules and modulate distinct cellular responses, including gene transcription, cell migration and survival, which can result in patient death Citation[3,4]. We recently found that cancer cells, including ovarian cancer cells, selected for their resistance to chemotherapy or isolated from metastatic sites, expressed high basal levels of tissue transglutaminase (TG2), a multifunction protein Citation[5–7]. In conjunction with earlier reports, we found that TG2 can associate with cell surface integrins in a 1:1 complex Citation[8–11] and promote cell adhesion and migration on fibronectin, implying that aberrant TG2 expression may play an important role in conferring drug resistance and the metastatic phenotype in ovarian cancer cells. Indeed, ectopic TG2 expression rendered ovarian cancer cells resistant to paclitaxel and promoted cell invasion through Matrigel™-coated transwell inserts, while TG2 downregulation by siRNA resulted in increased sensitivity to chemotherapy and attenuated invasion Citation[7]. In line with these observations, Satpathy et al. reported that increased TG2 expression in ovarian cancer cells enhanced cell adhesion to fibronectin and promoted directional cell migration Citation[12]. In addition, ovarian cancer cells with TG2 knockdown showed diminished tumor dissemination on the peritoneal surface and mesentery in an intraperitoneal ovarian xenograft mouse model. Importantly, in patients with ovarian cancer (n = 93), TG2 overexpression was associated with significantly worse overall survival in both univariate and multivariate analyses Citation[7]. These findings suggest that TG2 overexpression is an adverse prognostic factor in ovarian cancer and that targeting TG2 may be an attractive therapeutic approach.
TG2: the protein with a split personality
Tissue transglutaminase is structurally and functionally a complex protein with both intracellular and extracellular functions. In addition to catalyzing Ca2+-dependent transamidation reactions Citation[13,14], it can bind to and hydrolyze GTP/GDP with an affinity and catalytic rate similar to the α-subunit of large heterotrimeric G-proteins and Ras-type G-proteins Citation[15]. In addition, TG2 can serve as a protein disulfide isomerase and as a kinase Citation[13]. As a scaffold protein, TG2 can bind to and alter the structure and function of various proteins, including Akt, PTEN and hypoxia-inducing factor. Such TG2-induced alterations can impinge upon various aspects of cancer cells, including cell growth, survival and invasion Citation[5]. Although predominantly a cytosolic protein, TG2 can be secreted outside the cells, where it can cross-link and render the ECM proteins resistant to mechanical and proteolytic insults Citation[16]. TG2-mediated cross-linking of the ECM also plays an important role in the deposition and stabilization of the ECM, which, in turn, promotes cell attachment and spreading. TG2 can translocate to the cell membrane, where it binds tightly to both the cell surface integrins and fibronectin and promotes cell attachment, migration and invasion. With the help of importin-α3, TG2 can translocate to the nucleus, where it functions as a G-protein or alternatively cross-links such important proteins as histones, retinoblastoma protein and SP1 transcription factor Citation[17]. Another enigmatic feature of TG2 is that it can function as a proapoptotic and antiapoptotic protein. Interestingly, despite the variety of functions in which TG2 participates, TG2-knockout mice show no phenotype and are anatomically, developmentally and reproductively normal Citation[18].
TG2 in cancer cells
Several experimental observations support the hypothesis that TG2 plays a role in the progression of certain cancer types. First, multiple studies have demonstrated that the TG2 protein is upregulated in pancreatic carcinoma Citation[19], breast carcinoma Citation[20], malignant melanoma Citation[8], glioblastoma Citation[21] and ovarian carcinoma Citation[7,12]. In addition, a positive correlation between chemotherapy resistance and the metastatic potential of certain cancers and TG2 expression has been demonstrated Citation[5,6]. Finally, several tumor cell types with TG2 overexpression show resistance to apoptosis, while siRNA-mediated downregulation or treatment with TG2 inhibitors sensitized cancer cells to apoptosis Citation[7,9,21–25].
In a study to identify metastasis-associated proteins by proteomic analysis, Jiang et al. observed that TG2 was one of the 11 proteins selectively amplified in metastatic human lung carcinoma Citation[26]. Similarly, a comprehensive analysis of more than 30,000 genes using three different techniques revealed that TG2 was one of the most differentially expressed genes in pancreatic tumors Citation[27]. In addition, treating breast cancer cells with EGF induced TG2 expression and conferred resistance to doxorubicin-induced apoptosis Citation[28]. By contrast, there are also reports suggesting that TG2 expression is downregulated during the process of carcinogenesis. For example, Xu et al. reported that the GPR56 protein is downregulated in highly metastatic melanoma cancer cells and that TG2 is a binding partner for GPR56, suggesting that TG2 can act as a tumor-suppressing protein through its interaction with GPR56 Citation[29]. These paradoxical findings suggest that TG2 expression may depend upon the type, location and stage of the cancer. Therefore, precise understanding of TG2-regulated functions in relation to cancer stage and type is important for implementing TG2-based interventions to disrupt malignant cell invasion, growth and survival.
TG2 & cell signaling
Tissue transglutaminase can interact with various signaling molecules and modulate signaling pathways. outlines key pathways that can be modulated by TG2 to influence the growth, cell survival and invasion of cancer cells. For example, integrins, the cell surface receptors that promote cell attachment and communication with the ECM, can influence several aspects of cancer cell behavior, including motility, invasion, growth and survival Citation[30]. In general, integrins represent low-affinity receptors (1 × 106–1 × 109l/mol) and can only bind to ECM ligands only when the integrins exceed a certain critical number in the form of focal contacts or hemidesmosomes. However, certain cellular proteins can interact with and modulate the binding affinity of integrins. Such interaction has been observed between integrin β3 and the integrin-associated protein, also referred to as CD47 Citation[31]. Signaling transmitted by integrins inside the cells can be modulated by such protein–integrin interactions. In view of these observations and the observation that ovarian cancer cells express high basal levels of TG2 Citation[7,12], it is tempting to speculate that TG2 expression could modulate integrin-mediated signaling and affect cell growth, survival and metastasis. Indeed, TG2 is known to promote interactions between cell surface integrins and fibronectin to support cell adhesion, motility, invasion and survival functions Citation[8–11]. In addition, syndecan-4 (S4) can also interact with TG2-bound integrins, raising the possibility that TG2 contributes to cell adhesion by binding to S4. Thus, S4-null fibroblasts showed lower levels of focal adhesion kinase (FAK; Y397) during the initial phase of cell adhesion to fibronectin than wild-type cells. A similar defect was observed in early FAK (pY397) activation in TG2-null mouse embryonic fibroblasts compared with the wild-type fibroblasts; the early FAK activation was accompanied by hyperactivation of RhoA and decreased cell migration Citation[32].
There is direct evidence supporting the claim that TG2 overexpression in ovarian cancer cells promotes cell attachment, migration and resistance to chemotherapy Citation[7,12,24]. Thus, TG2 knockdown by siRNA significantly inhibited cell attachment, attenuated invasion and rendered cells sensitive to chemotherapy Citation[7,23]. Conversely, ectopic TG2 expression promoted cell attachment, invasion and resistance to chemotherapy drugs. TG2-mediated resistance was related to activation of the nuclear transcription factor NF-κB Citation[23] and the Akt pathways Citation[7]. In addition, TG2 can also regulate FAK activation. FAK is a 125-kDa nonreceptor protein tyrosine kinase that plays an important role in cell survival, migration and invasion. FAK-induced signaling is initiated at sites of integrin-mediated cell adhesions (or focal adhesions) and activates protein–protein interaction adaptor functions at sites of cell attachment to the ECM Citation[33]. TG2, associated with integrin β1, has been shown to promote FAK activation in response to fibronectin binding Citation[9,11,19,34]. TG2 can also activate FAK and its downstream Akt pathway as a result of its direct interaction with the FAK protein Citation[19], probably inducing conformational changes in FAK leading to FAK autophosphorylation (pY397). Once phosphorylated, FAK can recruit the Src kinase, which phosphorylates other tyrosine sites on FAK (Y407, Y576, Y577, Y861 and Y925), leading to the recruitment and activation of several downstream signaling proteins, such as RAS/ERK, PI3K/Akt and Crk/Dock180/Rac, which can, in turn, impact on cell growth, survival and invasion.
Further adding to the complexity of TG2 in FAK and Akt signaling is the observation that TG2 can modulate the expression of the tumor-suppressor protein PTEN Citation[35]. PTEN can inhibit cell growth, invasion, migration and focal adhesions primarily by dephosphorylation and inactivation of Akt and FAK. Deletion and mutation of the PTEN gene are observed frequently in various human cancers. These genetic changes, however, are not necessarily the only way to inactivate PTEN during tumorigenesis; some subtle changes induced by post-translational modifications can also affect the stability and degradation of PTEN, which can profoundly affect tumorigenesis. Indeed, a recent report demonstrated that the oncogenic protein NEDD4-1 acts in a similar manner to the E3 ubiquitin ligase for PTEN and promotes PTEN degradation through the ubiquitin–proteasomal pathway Citation[35]. Interestingly, TG2 overexpression in cancer cells is able to induce ubiquitination and proteasomal degradation of PTEN Citation[34]. Thus, TG2 can promote FAK/Akt activation in two ways: first, by inducing autophosphorylation of FAK; and second, by inhibiting the phosphatase activity of PTEN, resulting in constitutive activation of the FAK/Akt pathway.
In addition, TG2 expression in cancer cells is implicated in the constitutive activation of NF-κB, which plays an important role in regulating genes that are involved in cell growth, apoptosis and metastasis Citation[36]. Indeed, inhibition of TG2 activity by siRNA or a small-molecule inhibitor attenuated NF-κB activation and reversed the sensitivity of drug-resistant breast and ovarian cancer cells to doxorubicin and cisplatin, respectively Citation[7,9,24]. These results imply that TG2-induced activation of NF-κB plays a role in conferring drug resistance in cancer cells.
TG2 as a therapeutic target
From the preceding discussion, it is apparent that inhibition of TG2 could be a useful way of reversing chemotherapy resistance in cancer cells. In a previous study, we determined the therapeutic potential of elevated TG2 expression for treating orthotopically grown ovarian tumors. Using small (<100 nm), neutrally charged liposomes (dioleoyl-phosphatidylcholine [DOPC]) as the delivery system, we found that TG2 siRNA could effectively downregulate TG2 expression in orthotopically grown tumors. We found that treating mice with TG2 siRNA-DOPC alone or combined with docetaxel demonstrated significant antitumor activity against orthotopically grown chemotherapy-sensitive (HeyA8) and -resistant (HeyA8-MDR) ovarian tumors Citation[7]. To determine the potential mechanisms underlying the antitumor effect of TG2 siRNA-DOPC, we then examined its effect on several biological end points, including cell proliferation (Ki-67), angiogenesis (CD31) and apoptosis (TUNEL). A reduction of more than 90% in Ki-67 expression was evident (p < 0.001) in tumors obtained from mice administered TG2 siRNA-DOPC alone or combined with docetaxel. Tumors obtained from mice treated with docetaxel alone or control siRNA-DOPC consistently showed high levels of Ki-67 staining. We also evaluated the blood vessel density in tumors obtained from mice treated with control siRNA-DOPC, docetaxel alone, TG2 siRNA-DOPC alone or TG2 siRNA-DOPC plus docetaxel. The results revealed a significant decrease in the mean blood vessel density in tumors recovered from mice that received TG2 siRNA-DOPC or docetaxel alone. A densitometric analysis showed a significant difference in the mean microvascular density (p < 0.0013) in mice that received TG2 siRNA-DOPC plus docetaxel. Finally, we evaluated apoptosis in orthotopic tumors using TUNEL staining. In ovarian tumors, minimal cell apoptosis was apparent in the empty liposome, control siRNA-DOPC or control siRNA-DOPC with docetaxel-treatment groups. However, treatment with TG2 siRNA-DOPC alone and TG2 siRNA-DOPC plus docetaxel resulted in a significant increase in apoptosis (p = 0.027 and p = 0.001, respectively). Interestingly, the increase in apoptosis in the TG2 siRNA-DOPC plus docetaxel group was greater than in the TG2 siRNA-DOPC alone group (p = 0.004). In summary, targeted therapy with TG2 siRNA combined with docetaxel significantly reduced tumor growth in chemotherapy-sensitive and -resistant models. Given the clinical relationship between TG2 expression and the poor prognosis of patients with cancer, our findings raise the possibility that TG2 silencing combined with chemotherapy could be a promising therapeutic option.
In this regard, using siRNA to silence TG2 expression holds great promise for the development of therapeutics directed against drug-resistant and metastatic cancers. For example, using siRNA for gene silencing has rapidly become a powerful tool for determining protein function and gene discovery. In addition, the sequence specificity of siRNA makes it an attractive tool for cancer therapy. Moreover, siRNA-based therapeutics have the potential to inhibit molecular targets that are not treatable with conventional small molecules, antibodies or other biological agents. However, a major challenge in using siRNA-based drugs in humans is effectively delivering the drug to the target tissue, as naked siRNA is unstable in plasma. By contrast, chemically modified siRNA is thought to be relatively stable when administered systemically in mice but the siRNA must be administered in large doses (30–50 mg/kg). Such large doses of siRNA may not be practical for therapeutic applications in humans because of the costs and safety issues. However, encapsulation of siRNA in lipid-based nanoparticles of 100 nm or less in diameter provides effective dosing in mice and monkeys at low amounts (0.2–2 mg/kg). As a proof of concept, we showed that the use of DOPC nanoliposomes for delivering TG2 siRNA to orthotopically grown pancreatic Citation[38] and ovarian Citation[7] tumors in mice was effective in silencing the target gene and inhibiting tumor growth and metastasis. From these studies, it is apparent that using TG2 siRNA can be rapidly translated into the clinical setting for treating chemotherapy-resistant ovarian cancer.
Five-year view
Although ovarian cancer ranks seventh among all cancers in women in terms of prevalence, almost 60% of those with ovarian cancer eventually die of the disease. Drug resistance is thought to cause treatment failure and death in more than 90% of patients with metastatic disease. Therefore, understanding tumor-encoded genes, whose expression contributes to drug resistance and metastasis of ovarian cancer cells, may reveal novel therapeutic targets for effective control and treatment of the disease. TG2 is one such gene whose expression is elevated in chemotherapy-resistant cancer cells or cells isolated from metastatic sites. High levels of TG2 expression in ovarian tumor samples corresponded with significantly lower patient survival rates than in those with low levels of TG2 expression. Moreover, tumors with TG2 overexpression tended to have an increased ability to invade healthy tissue and to survive or avoid the effect of chemotherapy. These results suggest that silencing TG2 could be an effective way to overcome drug resistance in ovarian cancer. Indeed, drug-resistant (HeyA8-MDR), orthotopically grown tumors in a nude mouse model became responsive to chemotherapy when treated with TG2 siRNA. Thus, silencing TG2 by siRNA may provide a novel therapeutic approach for late-stage ovarian cancer.
Financial & competing interests disclosure
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Jemal A, Siegel R, Ward E et al. Cancer Statistics. CA Cancer J. Clin.57, 43–66 (2007).
- Mcguire WP, Hoskins WJ, Brady MF et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N. Engl J. Med.334, 1–6 (1996).
- Yamada KM, Miyamoto S. Integrin transmembrane signaling and cytoskelatal control. Curr. Opin. Cell Biol.7, 681–689 (1995).
- Ricciardelli C, Rodgers RJ. Extracellular matrix of ovarian tumors. Sem. Reproduc. Med.24, 270–282 (2006).
- Verma A, Mehta K. Tissue transglutaminase-mediated chemoresistance in cancer cells. Drug Resist. Update10, 144–151 (2007).
- Mangala SL, Gillogli M, Sahin S, Arun B, Mehta K. Tissue transglutaminase-mediated alterations in extracellular matrix inhibit invasion. Mol. Cancer4, 1–8 (2005).
- Hwang JH, Mangala LS, Fok JY et al. Clinical and biological significance of tissue transglutaminase in ovarian carcinoma. Cancer Res.68, 5849–5858 (2008).
- Fok JY, Ekmekcioglu S, Mehta K. Implications of tissue transglutaminase expression in malignant melanoma. Mol. Cancer Ther.5, 1493–1503 (2006).
- Herman JF, Mehta K. Implications of increased tissue transglutaminase expression in drug-resistant breast cancer (MCF-7) cells. Oncogene25, 3049–3058 (2006).
- Mangala LS, Fok JY, Zorrilla-Calancha IR, Verma A, Mehta K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene26, 2450–2470 (2007).
- Akimov SS, Krylov D, Fleishmann LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol.148, 825–838 (2000).
- Satpathy M, Cao L, Pincheira R et al. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res.67, 7194–7202 (2007).
- Lorand L, Graham R. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat. Mol. Cell Biol.4, 140–156 (2003).
- Mehta K. Transglutaminases: family of enzymes with diverse functions. Prog. Exp. Tumor Res.38, 1–18 (2005).
- Begg GE, Holman SR, Stokes PH et al. Mutation of a critical GTP-binding arginine in transglutaminase 2 disinhibits intracellular crosslinking activity. J. Biol. Chem.281, 12603–12609 (2006).
- Verderio EAM, Johnson TS, Griffin M. Transglutaminases in wound healing and inflammation. Prog. Exp. Tumor Res.38, 89–114 (2005).
- Ballestar E, Boix-Chornet M, Franco L. Conformational changes in the nucleosome followed by the selective accessibility of histone glutamines in the transglutaminase reaction; effect of salt concentrations. Biochemistry40, 1922–1929 (2001).
- Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J. Biol. Chem.276, 20673–20678 (2001).
- Verma A, Wang H, Manavathi B et al. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res.66, 10525–10533 (2006).
- Mehta K, Fok J, Miller FR, Koul D, Sahin AA. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin. Cancer Res.10, 8068–8076 (2004).
- Yuan L, Siegel M, Choi K et al. Transglutaminase 2 inhibitor, KCC009 disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene26, 2563–2573 (2006).
- Akar A, Ozpolat B, Mehta K, Fok JY, Kondo Y, Lopez-Berestein G. Tissue transglutaminase (TG2) expression protects pancreatic cancer cells from autophagy. Mol. Cancer Res.5, 241–249 (2007).
- Kim DS, Park SS, Nam BH, Kim IH, Kim SY. Reversal of drug resistance in breast cancer cells by transglutaminase 2 inhibition and nuclear factor-κB inactivation. Cancer Res.66, 10936–10943 (2006).
- Cao L, Petrusca DN, Satpathy M et al. Tissue transglutaminase protects epithelial ovarian cancer cells from cisplatin induced apoptosis by promoting cell survival signaling. Carcinogenesis29, 1893–1900 (2008).
- Verma A, Mehta K. Tissue transglutaminase-mediated activation of nuclear transcription factor-κB as a target for cancer treatment. Curr. Cancer Drug Targets7, 559–565 (2007).
- Jiang WG, Ablin R, Douglas-Jones A, Mansel RE. Expression of transglutaminases in human breast cancer and their possible clinical significance. Oncol. Rep.10, 2039–2044 (2003).
- Iacobuzio-Donahue CA, Ashfaq R, Maitra A et al. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res.63, 8614–8622 (2003).
- Antonyak MA, Miller AM, Jansen JM et al. Augmentation of tissue transglutaminase expression and activation by epidermal growth factor inhibit doxorubicin-induced apoptosis in human breast cancer cells. J. Biol. Chem.279, 41461–41467 (2004).
- Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase and inhibits melanoma tumor growth and metastasis. Proc. Natl Acad. Sci. USA103, 9023–9028 (2006).
- Felding-Habermann B. Integrin adhesion receptors in tumor metastasis. Clin. Exp. Metastasis20, 203–213 (2003).
- Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol.11, 130–135 (2001).
- Kotsakis P, Griffin M. Tissue transglutaminase in tumour progression: friend or foe? Amino Acids33, 373–384 (2007).
- McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal adhesion kinase in cancer – a new therapeutic opportunity. Nat. Rev. Cancer5, 505–515 (2005).
- Verma A, Koul D, Guha S, Wang H, Mehta K. Transglutaminase-mediated regulation of PTEN in pancreatic cancer cells. Clin. Cancer Res.14, 1997–2005 (2008).
- Wang X, Trotman LC, Koppie T et al. NEDD4–1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell128, 129–139 (2007).
- Karin M, Cao Y, Greten F, Li Z. NF-kB in cancer: from innocent bystander to major culprit. Nature Rev. Cancer2, 301–310 (2002).
- Mann AS, Verma A, Wang H et al. Overexpression of tissue transglutaminase leads to constitutive activation of nuclear factor-κB in cancer cells: delineation of a novel pathway. Cancer Res.66, 8788–8795 (2006).
- Verma A, Guha S, Diagaradjane P et al. Tissue transglutaminase as a therapeutic target for inhibiting growth and metastasis of pancreatic ductal adenocarcinoma. Clin. Cancer Res.14, 2476–2483 (2008).