Abstract
T lymphocytes are the primary targets of immunotherapy in clinical transplantation; however, B-lymphocytes and their secreted alloantibodies are also highly detrimental to the allograft. Therefore, the achievement of sustained organ transplant survival will likely require the induction of B-lymphocyte tolerance. During development, acquisition of B-cell tolerance to self-antigens relies on clonal deletion in the early stages of B-cell compartment ontogeny. We contend that this mechanism should be recapitulated in the setting of alloantigens and organ transplantation to eliminate the alloreactive B-cell subset from the recipient. Clinically feasible targets of B-cell-directed immunotherapy, such as CD20 and B-lymphocyte stimulator (BLyS), should drive upcoming clinical trials aimed at remodeling the recipient B-cell repertoire.
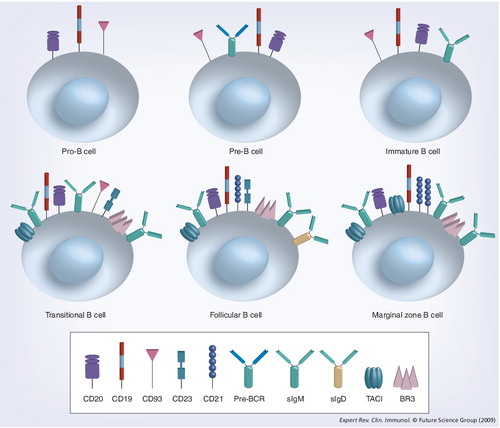
BCR: B-cell receptor; BR3: B-lymphocyte stimulator receptor 3; TACI: Transmembrane activator 1 and calcium-signaling modulator and cyclophilin ligand-interactor.
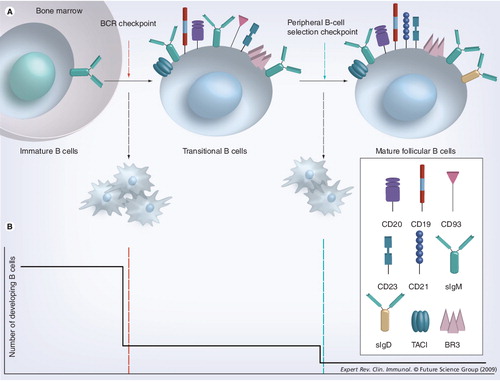
(A) Immature B cells have successfully rearranged their heavy and light chain immunoglobulin genes. During the BCR checkpoint, immature B cells that are engaged by high avidity antigens are induced to die, while those who do not, transit to the periphery and become transitional B cells. The continued survival of transitional B cells in the periphery is dictated by the availability of B-lymphocyte stimulator (BLyS) and relative BCR tonic signaling. (B) The frequency of developing B-cell survival is depicted here as ontogeny progresses across the BCR and peripheral selection checkpoints. At the BCR checkpoint (red line), only 10% of the immature B cells survive, while at the peripheral B-cell selection checkpoint (green line), approximately 30% of the remaining B cells become mature follicular B cells.
BCR: B-cell receptor; BR3: B-lymphocyte stimulator receptor 3; TACI: Transmembrane activator 1 and calcium-signaling modulator and cyclophilin ligand-interactor.
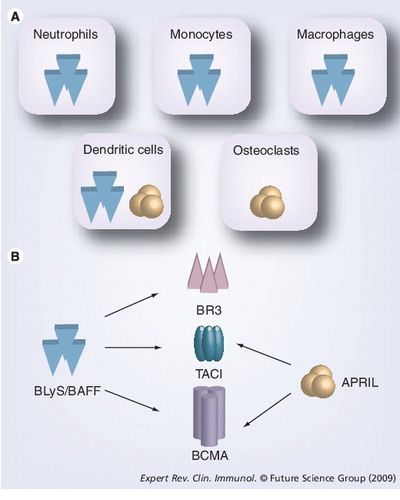
(A) Cells producing BLyS and/or APRIL. (B) Relative binding affinities for each receptor–ligand combination are very similar. However, it is important to note that the primary BLyS receptor responsible for regulating pre-immune B-cell homeostasis is BR3.
APRIL: A proliferation-inducing ligand; BAFF: B-cell-activating factor; BCMA: B-cell maturation antigen; BLyS: B-lymphocyte stimulator; BR3: B-lymphocyte stimulator receptor 3; TACI: Transmembrane activator 1 and calcium-signaling modulator and cyclophilin ligand-interactor.
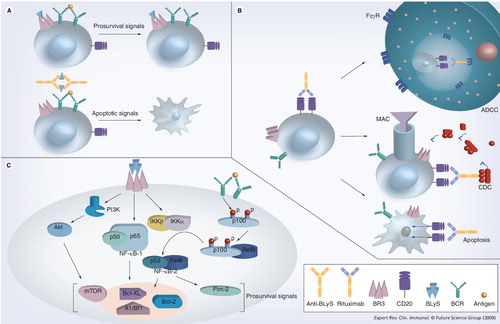
(A) BLyS–BR3 binding and tonic BCR signaling promotes B-cell survival, while anti-BLyS treatment favors apoptotic signals. (B) Anti-CD20 (e.g., rituximab) causes B-cell death via three separate mechanisms. (C) Prosurvival signals are upregulated in the setting of downstream cross-talk after BCR crosslinking and BLyS–BR3 binding.
ADCC: Antibody-depedent cell-mediated cytotoxicity; BCR: B-cell receptor; BLys: B-lymphocyte stimulator; BR3: B-lymphocyte stimulator receptor 3; CDC: Complement-dependent cytotoxicity; MAC: Membrane-attack complex.
(A) After standard immunotherapy, the alloreactive B-cell subset persists, producing donor-specific antibodies, and ultimately causing chronic allograft rejection. (B) In the B-cell repertoire remodeling scenario, the alloreactive B-cell subset of the pre-immune B-cell repertoire is eliminated after B-cell depletion therapy. Induction T-cell depletion therapy is also given. Following transplantation, the B-cell repertoire undergoes reconstitution in the presence of alloantigens permitting clonal deletion of the alloreactive subset under B-lymphocyte stimulator-limiting conditions. In the absence of donor-specific B cells, allograft survival may improve.
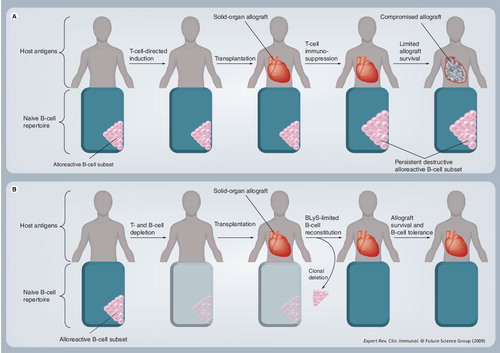
(A) After rituximab induction therapy, B cells are depleted and BLyS levels rise, risking the development of alloreactive B cells upon reconstitution. (B) A similar scenario occurs with anti-BLyS induction. (C) Maintenance anti-BLyS following B-cell depletion creates BLyS-limiting conditions that minimize the risk of developing alloreactive B cells and antibody-mediated rejection.
BCMA: B-cell maturation antigen; BLyS: B-lymphocyte stimulator; BR3: B-lymphocyte stimulator receptor 3; GC: Germinal center; TACI: Transmembrane activator 1 and calcium-signaling modulator and cyclophilin ligand-interactor.
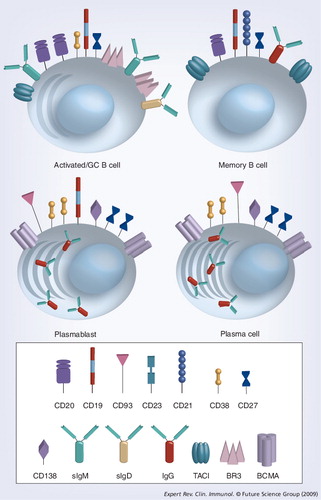
A myriad of immunologic barriers challenge the short- and long-term success of solid organ transplantation. A major priority for the field of transplantation is to identify nontoxic, non-neoplastic immune-modulating methods capable of overcoming these barriers and promoting a sustained state of donor-specific immunological tolerance. Immunological tolerance is defined as the absence of an immune response against any particular antigen. In the setting of transplantation, this term is used to describe tolerance to alloantigens (primarily polymorphisms at the HLA locus). It is a well-established principle that immune tolerance is achieved either through unresponsiveness of reactive cells or their clonal elimination from the lymphocyte repertoire. The detailed study of the mechanisms governing tolerance to self-antigens over the past century has provided invaluable insights necessary for the establishment of alloimmune tolerance in the clinical setting.
Many strategies have attempted to curtail delayed graft function and eliminate episodes of acute rejection by promoting alloimmune tolerance. To date, these protocols have been primarily directed at the T-cell compartment. Immunosuppression at the time of transplantation (induction) and following transplantation (maintenance) effectively combats the upregulated immunogenic molecules that circulate in the brain-dead donor and reduces the incidence of rejection Citation[1–3]. Induction therapy with rabbit antithymocyte globulin (thymoglobulin) in combination with immunosuppressive therapy causes a reduced incidence and severity of acute rejection in adult renal transplant recipients when compared with regimens without induction thymoglobulin Citation[1,4]. Numerous protocols, especially in Europe, have been designed to customize immunosuppressive protocols to individual patients’ risk factors in order to achieve donor-specific tolerance. Many of the current strategies also highlight calcineurin inhibitor-sparing protocols, mammalian target of rapamycin-inhibitor protocols and corticosteroid-limiting protocols to minimize toxicities and induce greater alloimmune tolerance Citation[5].
A strategy that found initial success was the practice of lymphoid irradiation and thymoglobulin induction therapy after combined kidney and hematopoietic stem cell transplantation from HLA-matched donors Citation[6,7]. The continued presence of donor immune cells (chimerism) in the recipient’s thymus and peripheral lymphoid tissue promoted alloimmune tolerance by eliminating T-cell clones specific for graft-derived alloantigens Citation[8]. Despite the successes of these T-cell tolerance-promoting strategies, B-cell tolerance in transplantation remains a challenge Citation[9]. Indeed, B-cell immunity was found to persist after combined kidney and bone marrow transplantation in the setting of non-myeloablative conditioning that produced T-cell tolerance Citation[10]. Two out of the four patients in this study developed de novo donor-specific HLA antibodies, which were preceded by elevated serum B-lymphocyte stimulator (BLyS) levels Citation[10].
In fact, the last decade has witnessed a growing body of evidence implicating B cells as key participants in alloimmunity Citation[11–18]. The B-cell response to alloantigen, quantified by the development of donor-specific alloantibodies and histopathological evidence of antibody-mediated rejection (AMR), has been shown to poorly impact the long-term survival of organ allografts and patient survival rates Citation[9,16,19,20]. For example, 1 year post-transplantation, allograft failure rates were higher among those who developed donor-specific antibodies (DSAs) compared with those who did not (6.6 vs 3.3%; p = 0.0007) Citation[21]. Furthermore, in a recent investigation at 5.5 years after renal transplantation, the presence of DSAs was associated with a significantly lower graft survival (49 vs 83% in the HLA antibody-negative group) Citation[22]. Also, the impact of non-DSAs on graft survival was poor (70 vs 83%; p = 0.0001) Citation[22]. During acute cellular and antibody-mediated rejection, the development of donor-specific alloantibodies is evident as either diffuse or focal C4d deposition on renal allograft tissue biopsy Citation[23,24]. A germinal center reaction, the inciting event, occurs when mature follicular B cells encounter both alloantigen and sufficient T-cell help (in the form of CXC chemokine ligand 13 and the adaptor protein signaling lymphocyte-activation molecule, among other chemokines), and as a result undergo proliferation and subsequently differentiate into plasma cells Citation[25–29].
In studies of sera collected from patients who rejected their renal allograft, 86–96% developed alloantibodies before their graft failure occurred Citation[9,30]. The presence of DSAs in patients with a functioning allograft (heart, lung, kidney and liver) is quite common (22.8, 14.2, 21–23 and 19.3%, respectively) Citation[21,31]. Although the appearance of DSAs portends a poor prognosis, the development of allograft rejection occurs at a variable rate Citation[30]. For example, the presence of alloantibodies within 1 year of transplantation has a mean time to graft failure of 5.1 years, while alloantibodies that form after 1 year have a slower rate of rejection (80% survival at 10 years and 50% survival at 15 years after transplantation) Citation[9,30]. Overall, it is our contention that the establishment of robust B-lymphocyte tolerance to organ allografts is a requisite step in the achievement of sustained transplantation tolerance.
What would B-cell tolerance in transplantation involve?
We define B-cell tolerance as the absence of alloreactive B cells and/or DSAs, which typically form after the initiation of the germinal center reaction to alloantigen. Several regimens for achieving B-cell tolerance are available, each having different mechanisms and potential pitfalls. For example, alemtuzumab (campath-1H, a humanized anti-CD52 monoclonal antibody) depletes T-cells, B cells, natural killer cells, and other lymphoid cells, and has shown good long-term allograft survival rates (similar to thymoglobulin) Citation[32,33]. However, despite these results, true B-cell tolerance has been elusive, as patients continue to develop DSAs and can develop AMR Citation[34,35]. By contrast, donor-specific B-cell tolerance was demonstrated dramatically in infants who underwent ABO-incompatible heart transplantation Citation[36]. This work illustrates the importance of exploiting the immature immune system for its inherent malleability Citation[37]. Furthermore, although relative B-cell tolerance after renal transplantation has been reported with blockade of the costimulatory interaction between CD40 and CD154 in nonhuman primates, the effect may be lost after the treatment is stopped Citation[38]. Efforts to produce B-cell tolerance in alloantigen-naive adult patients (as opposed to recipients presensitized with donor-specific alloantibodies or recipients with AMR) have been limited. The only randomized, controlled study to date was suspended after five out of six renal allograft recipients who received B-cell-directed induction therapy with rituximab developed acute cellular rejection, demonstrating that new strategies must be taken to achieve B-cell tolerance Citation[39].
In this perspective, we will address the question of how B-cell tolerance in alloantigen-naive transplantation might be possible. We contend that the achievement of sustained allograft survival will require not only T-cell-depleting immunotherapy (such as thymoglobulin) but also immune intervention strategies to modulate the alloreactive B-cell response. We will review the fundamental mechanisms that govern B-cell self-tolerance, since their application in clinical B-cell alloimmune tolerance formation is a biological necessity. Following this review, we will present our argument for how B-cell tolerance in transplantation might be pursued and discuss what challenges face its realization in alloantigen-naive and -presensitized patients.
Overall, B-lymphocytes are now increasingly recognized as key participants in the alloimmune response Citation[11,12]. The mechanisms governing long-lived plasma cell (LLPC) homeostasis after transplantation are not well understood, highlighting the complexity of antigen-experienced B cells and the corresponding antibody-mediated rejection Citation[40]. Given this clinical challenge, a persuasive argument could be made in favor of B-cell-targeted therapy at the time of transplantation Citation[41]. This strategy may promote the purging of alloreactive B-cell clones, the repopulating of the pre-immune B-cell pool with non-alloreactive specificities Citation[42], and potentially limit the development of allo-AMR.
B-cell ontogeny
B cells are generated continuously in the bone marrow, with their production rate declining progressively throughout life Citation[43]. Derived from hematopoietic stem cells, B cells pass through a series of steps that involve the rearrangement of their immunoglobulin (Ig) genes, culminating in B-cell antigen receptor expression. B cells and their secreted antibodies have an extremely diverse spectrum of antigen-recognition specificities (referred to as the B-cell ‘repertoire’) for which pathogens can be bound and effectively neutralized. The factors controlling the primary (‘naive’) B-cell repertoire size and clonotypic composition are of fundamental importance, as these are the cells from which antibody-forming cells and long-lived memory cells arise Citation[42].
Bone marrow B-cell developmental stages are defined according to two general nomenclatures Citation[44–48]. The first, forwarded by Osmond Citation[47] and Melchers Citation[48], delineates pro-B, pre-B and immature B-cell stages of differentiation according to the rearrangement status of the IgH and IgL chain genes. The second, attributed to Hardy Citation[49], uses lettered categories (A–E) to further divide developing B cells according to their proliferative status and surface markers. In the pro-B-cell stage, which corresponds generally to Hardy Fractions A–C, cells committed to the B lineage undergo D–JH and VH-DJH gene rearrangements Citation[50]. A successful IgH rearrangement leads to the expression of surrogate light chains and surface expression of the pre-B-cell antigen receptor. These cells, termed ‘Hardy Fraction C’, undergo a proliferative burst then enter the pre-B-cell stage (Hardy Fraction D) . The light chain gene rearrangement ensues and, if successful, yields the expression of a complete B-cell antigen receptor. This marks the immature bone marrow stage (Hardy Fraction E) . Immature B cells exit to the peripheral circulation and spleen as transitional B cells, where they undergo further differentiation before entering one of the mature, pre-immune B-cell pools. Most developing cells enter the follicular subset, which constitutes the bulk of the recirculating pre-immune B cells. Some cells join the marginal zone subset instead. The factors determining follicular versus marginal zone B-cell differentiation are still debated.
Pre-immune B-cell development & selection: balancing of homeostasis & tolerance
The naive mature B-cell pool is continuously subjected to mechanisms that maintain sufficient B-cell numbers for adequate immunological surveillance, while at the same time limiting self-reactivity and maximizing clonal diversity. The processes that govern B-cell tolerance to self-antigens will likely be the same as those that govern B-cell tolerance to alloantigens Citation[51]. Akin to tolerance to self-antigens, B-cell depletion therapy will permit the reconstituting B-cell pool to develop in the presence of the organ allograft, thereby allowing B-cell selection and tolerance to alloantigens. The mechanisms permitting B-cell tolerance to alloantigens are clonal deletion, receptor editing, clonal anergy and extrinsically mediated selection Citation[52].
Support for the idea that B-cell tolerance is imposed during development has accumulated over the last four decades Citation[53]. In 1975, Nossal and Pike forwarded the notion of deletional mechanisms, or the ‘clonal abortion theory’, as the basis for immune tolerance Citation[54]. This concept was supported by a variety of in vitro studies demonstrating that immature B cells were susceptible to death, rather than activation, upon B-cell antigen receptor engagement Citation[55]. In 1989, both the Nemazee and Goodnow laboratories presented elegant transgenic models demonstrating deletional tolerance in vivoCitation[56,57]. In both of these, immature B cells with transgenic B-cell antigen receptors undergo deletion when on genetic backgrounds containing the relevant autoantigen. Clonal deletion was also seen at the pro-/pre-B-cell stage when autoreactive B cells were reconstituted in sublethally irradiated hosts Citation[58]. The deletion of self-reactive B cells was also demonstrated in transgenic mice with peripherally expressed antigen Citation[59]. In 1994, Goodnow et al. found that elimination of self-reactive B cells occurs as the pre-immune B-cell repertoire competes for limiting follicular resources (subsequently identified as BLyS, see later) Citation[60]. Goodnow et al. also went on to demonstrate clonal deletion is not only evident in pre-immune repertoire selection but also in the antigen-experienced B cells Citation[61]. Importantly, antigen-induced negative selection can also help clear the primary repertoire of self-reactive clones Citation[62].
During their ontogeny, B cells transit several selection ‘checkpoints’ (i.e., defined moments in a cell’s development where its ongoing survival is determined) prior to entering mature, pre-immune pools Citation[42,43,63]. The first B-cell antigen receptor-related checkpoint occurs during heavy chain rearrangement. Upon successful VH-DJH rearrangement, cells express a so-called pre-B-cell antigen receptor, comprised of m heavy chain with a surrogate light chain containing the λ5 and V-pre-B1 and V-pre-B2gene products. This cell surface molecule is associated with the Igα–Igβ signaling heterodimer and thus has signaling capacity Citation[64]. Signals via the pre-B-cell antigen receptor are required for continued differentiation, thus cells that fail to successfully rearrange their heavy chain genes die. Whether cells bearing heavy chains with the propensity for autoreactivity are also eliminated at this point is debated, although some evidence suggests that this may occur based on complementarity-determining region 3 length or avidity. Cells that pass this checkpoint join the pre-B-cell pool, rearrange their light chain genes and express a complete B-cell antigen receptor as surface IgM. At this and all subsequent differentiative stages, selection based on B-cell antigen receptor specificity occurs. At each of these checkpoints, substantial cell losses are incurred as clones bearing potentially self-reactive, pathogenic or ineffective B-cell antigen receptor specificities are purged from the emerging repertoire. Fewer than 10% of immature B cells generated in the bone marrow actually exit and reach the transitional B-cell stages Citation[65], and two thirds of the remaining cells are lost during peripheral B-cell selection prior to final maturation . In general, the immature B cells whose B-cell antigen receptor is engaged with high avidity are induced to die or undergo secondary light chain rearrangements by receptor editing Citation[66–70].
In contrast to these mechanisms of tolerance engendered by high avidity B-cell antigen receptor interactions in the bone marrow, sustained low avidity interactions foster death during the transitional B-cell stages in the periphery. Therefore, an important distinction between the immature and transitional checkpoints is the acquisition of the ability for selection to be modified based on homeostatic demands. Thus, while losses at the immature checkpoint appear to be absolute and based solely on the B-cell antigen receptor signal strength, the stringency of selection at the peripheral checkpoint can be relaxed or strengthened based on the perceived systemic need for B cells. This relationship is mediated by the BLyS family of cytokines and receptors, through integrated signaling cross-talk with the B-cell antigen receptor at the translational stages and beyond (see later) Citation[71].
Self-reactive B cells entering the periphery may also become tolerant when they achieve a state of anergy, in which their B-cell antigen receptor is downregulated and IgD receptor signaling is limited Citation[72]. In 1991, Weigert et al. demonstrated that self-reactive B cells specific for DNA in transgenic mice were developmentally arrested Citation[73]. Other mechanisms for B-cell tolerance induction to self are the absence of T-cell help for T-cell dependent (TD) responses, deviation of the Th immune response and inhibitory regulatory T-cells.
BLyS family of TNF receptors & ligands
Owing to its central role in the homeostatic control of primary B cells, the BLyS family of TNF receptors and ligands has garnered considerable investigation over the past decade and is the topic of extensive reviews and commentaries Citation[42,43,63,74]. Multiple groups identified BLyS and published its sequence. Hence, BLyS is also referenced in the literature as B-cell-activating factor (BAFF). Two receptors for BLyS were initially identified: transmembrane activator 1 and calcium-signaling modulator and cyclophilin ligand-interactor (TACI) and B-cell maturation antigen (BCMA) . These receptors were found to also bind a proliferation-inducing ligand (APRIL; another member of the TNF family of ligands). TACI and BCMA knockouts did not demonstrate depletion of mature B cells, prompting further investigation.
A third BLyS receptor was identified by different groups, who coined the terms BLyS receptor 3 (BR3) and BAFF receptor Citation[75]. BR3-blocked mice resembled the A/WySnJ strain phenotype in which the transitional compartment is expanded and the primary follicular compartment is depleted Citation[76]. Further studies demonstrated that BLyS is the key survival factor for the mature follicular B-cell subset Citation[77–80] and that BR3 is considered the primary receptor for BLyS Citation[63]. Of note, the BR3-deficient human homolog has been described in common variable immunodeficiency patients Citation[81]. Similar to A/WySnJ mice, B-cell development is arrested at the transitional B-cell stage, causing a paucity of mature B cells and low IgG and IgM levels. By contrast, BLyS-overexpressing transgenic mice demonstrate B-cell hyperplasia and numerous autoimmune diseases Citation[77,82].
During B-cell ontogeny, the immature CD23- fraction of B cells demonstrates minimal BLyS binding, while somewhat higher BLyS binding is seen in the more developed CD23+ immature fraction Citation[83]. As immature cells exit the bone marrow, their BLyS binding capacity increases, as surface expression of BR3 and TACI increases Citation[83]. In the periphery, critical interactions between B-cell antigen receptor signaling and BR3 expression have been observed. B-cell antigen receptor signaling regulates BR3 levels in late translational and mature follicular B cells Citation[76]. Via downstream cross-talk, B-cell antigen receptor signaling supplies an NF-κB substrate, p100, for BR3-mediated processing and B-cell survival beyond the transitional 1 stage Citation[71]. BLyS stimulation promotes NF-κB activation and also upregulates a variety of antiapoptotic proteins, including A1/Bfl-1, Bcl-xL and Bcl-2 Citation[84,85]. Other prosurvival signals that BLyS-BR3 binding causes include activation of Akt/mammalian target of rapamycin and Pim2 Citation[86].
Similar to other TNF receptors, BCMA, TACI and BR3 form trimeric complexes in vivoCitation[42]. The BLyS family of TNF receptors and ligands have similar but distinct affinities that account for their different functions. For example, although the affinities for BLyS with its three receptors are nearly identical, there are subtle differences . TACI and BCMA bind BLyS with decreasing affinity, respectively. In addition, TACI binds APRIL with equivalent strength and has a 100-fold greater affinity for APRIL than BLyS Citation[87–89]. APRIL is also known to bind heparin sulfate proteoglycans (e.g., syndecan-1/CD138) found on the surface of plasma cells (among other cells) Citation[90]. Importantly, BR3 only binds BLyS and not APRIL Citation[75]. As mentioned previously, BR3–BLyS interactions are vital to the maintenance of primary B cells, as knock-out and mutant mice clearly demonstrate Citation[91,92]. BR3 contains one extracellular cysteine-rich domain that binds BLyS and has a single intracellular recognition site for TRAF3 Citation[93]. The soluble BLyS trimer is generally considered the only active form and is produced by several different-cell types, including neutrophils, dendritic cells (DCs), synovial fibroblasts, astrocytes, nurse-like cells and T-cells, as well as in response to certain inflammatory mediators Citation[42].
A strategy towards the achievement of B-cell tolerance in alloantigen-naive recipients
Given the insights gained into the mechanisms of B-cell tolerance to self-antigens, we contend that B-cell alloantigen tolerance may be possible. We propose a regimen that ‘remodels’ the pre-immune B-cell repertoire of the alloantigen-naive patient . We define remodeling as a method of obtaining B-cell tolerance in which the alloreactive pre-immune B cells are eliminated at the time of induction – prior to transplantation. A new B-cell repertoire is permitted to repopulate in the presence of the allograft under BLyS limiting conditions (see below). Remodeling of the primary (nonsensitized) B-cell repertoire provides a potential novel approach for the promotion of B-cell tolerance in transplantation. Remodeling with alloantigen-driven negative selection for the induction of B-cell tolerance in transplantation would require the two key events, described here.
1. Allow the B-cell repertoire to reconstitute in the presence of the allograft
B-cell-targeted therapy would deplete the mature B cells and allow tolerance-promoting transitional B cells to reconstitute the peripheral B-cell compartment in the presence of the allograft Citation[41,94,95]. This strategy requires clonal deletion of alloreactive clones and subsequent replacement of these cells with nonalloreactive clones that identify the alloantigens as a component of the self-antigen repertoire. The most striking support for this concept was presented when ABO-incompatible infant heart transplant recipients demonstrated durable B-cell tolerance Citation[37]. West et al. observed that studies up to 8 years after ABO-incompatible infant heart transplant found a persistent selective deficiency of circulating donor-specific B cells and alloantibodies, and an absence of antibody-mediated damage Citation[36,37]. Animal models have demonstrated that persistent presence of the allograft can promote transplantation tolerance Citation[96,97]. Likewise, as mentioned earlier, self-reactive B cells from Ig transgenic mice were eliminated by clonal deletion or receptor editing after repeated exposure to antigen-bearing cells Citation[98]. Akin to the concept of B-cell repertoire remodeling, mixed allogeneic (or xenogeneic) bone marrow chimerism has been shown to permit B-cell tolerance and may have the potential for circumventing the hyperacute rejection associated with xenotransplantation Citation[8,99,100].
Evidence for the efficacy of B-cell targeting has also been shown in the nonhuman primate model of islet transplantation Citation[39,41]. Induction immunotherapy with rabbit antithymocyte globulin and rituximab (a humanized monoclonal antibody against the target CD20) promoted markedly prolonged islet allograft survival in nonhuman primates Citation[41]. Importantly, phenotypic analysis of the reconstituting B-cell compartment in monkeys with long-term graft survival demonstrated a predominance of tolerance-susceptible transitional B cells Citation[94], while hosts that rejected islet grafts had a larger proportion of mature follicular B cells Citation[41]. Furthermore, emerging results in the setting of murine islet allografts suggests that anti-BLyS induction and maintenance therapy can promote prolonged allograft survival (C57BL/6 → BALB/cJ MST >251 days; and BALB/cJ → C57BL/6 MST >168 days) Citation[95]. In these studies, a short course of rapamycin in conjunction with anti-BLyS treatment led to a predominance of tolerance-susceptible transitional B cells in vivo, which corresponded to marked CD4 T-cell hyporesponsiveness upon in vitro stimulation [Parsons RF, Yu M, Vivek K et al., Unpublished data] Citation[95].
Of note, no clinical investigation to date has examined the efficacy of anti-T-cell depletion therapy (e.g., with thymoglobulin) combined with B-cell depletion therapy toward improved allograft survival in alloantigen-naive patients. The need for investigations into B-cell tolerance regimens could not be more urgent. For example, an elegant study by Terasaki et al. has demonstrated that 23% of 1329 solid organ transplant recipients across 21 different centers who did not have preformed HLA antibodies at the time of transplantation did ultimately develop anti-HLA antibodies within 4 years of transplantation Citation[101]. Importantly, this study also found that those recipients who did develop anti-HLA antibodies had a significantly worse allograft survival rate versus those who did not form anti-HLA antibodies (58 vs 81%; p < 0.0001 after deceased donor transplantation, and 62 vs 78%; p < 0.0008 after living donor transplantation) Citation[101].
Interestingly, one recent randomized, clinical investigation did attempt B-cell induction therapy with rituximab and standard maintenance immunosuppression, and demonstrated a lack of protection against acute rejection of renal allografts Citation[39]; however, in this study T-cell induction therapy was not used in conjunction with rituximab induction treatment. Despite maintenance therapy with tacrolimus and mycophenolate mofetil, the rate of rejection is highly suggestive of T-cell-mediated rejection, underscoring the importance of a combined T–B-cell induction therapy approach. Further study is warranted to determine if the polarized proinflammatory cytokine profile observed in this study can be abrogated by a more balanced T–B-cell induction strategy. In addition, the consequence of regulatory B-cell depletion in these recipients should be determined, as our understanding of regulatory B cells is still emerging and several unknowns remain. The basic immunology literature to date argues that the downregulatory effects of regulatory B cells are derived from antigen-driven activation (e.g., with in vitro lipopolysaccharide or CD40 stimulation) and are primarily mediated by IL-10 secretion Citation[102,103]. Interestingly, several of the recipients who developed acute rejection after rituximab induction therapy actually had elevated levels of IL-10 (a cytokine with pro- and anti-inflammatory properties) Citation[104]. Hence, these data suggest any loss of the downregulatory effects of IL-10-secreting-regulatory B cells was well compensated for by the remaining lymphocytes. More basic investigation is needed for us to know exactly how B-cell depletion and subsequent reconstitution would impact the regulatory B-cell population.
2. Allow the B-cell repertoire to reconstitute without excess BLyS: permitting stringent transitional B-cell selection
The importance of BLyS levels for B-cell homeostasis and selection has been well established Citation[42]. Exogenous BLyS administration in vivo rapidly but reversibly doubled the peripheral B-cell pools Citation[79], while soluble BLyS-binding TACI Ig treatment markedly depleted most peripheral B-cell populations Citation[105]. Implications from these studies are that BLyS levels dictate the threshold of negative selection at the transitional à follicular B-cell peripheral tolerance checkpoint Citation[106,107]. In the presence of excess BLyS, self-reactive B cells are rescued from peripheral deletion and are permitted to enter the so-called ‘forbidden’ mature B-cell and marginal zone subsets Citation[106]. Furthermore, when the transitional B-cell compartment is lymphopenic, as in the nonobese diabetic mouse model of autoimmune diabetes, the stringency of negative selection at this checkpoint is lost and autoreactive B-cell clones escape to the periphery Citation[108]. Importantly, neutralization of circulating BLyS (via treatment with anti-BLyS antibody) restores the stringency of the transitional → follicular B-cell checkpoint, delaying the onset of Type 1 diabetes Citation[109]. However, in order to preserve the stringency of negative selection, a maintenance regimen of anti-BLyS antibody treatment was required to avoid a rebound increase in BLyS levels Citation[109]. Likewise, BLyS and APRIL neutralization has demonstrated a blunting of autoimmune activity and the prevention of Type 1 diabetes and antiphospholipid syndrome Citation[110,111]. Furthermore, as mentioned previously, the neutralization of BLyS in the context of murine islet transplantation promoted prolonged allograft survival and a 3-month period of tolerance-susceptible transitional B cells Citation[95]. These data indicate that BLyS neutralization warrants further investigation as a novel strategy for promoting transplantation tolerance.
The clinical studies of patients with systemic lupus erythematosis, rheumatoid arthritis and Sjögren’s syndrome have reported elevated BLyS levels and numerous autoreactive B-cell clonotypes. Accordingly, targeting BLyS with the fully humanized monoclonal antibody, belimumab, has shown tremendous promise for autoimmune diseases (as have other agents targeting B cells, such as rituximab and an anti-CD22 antibody, epratuzumab) Citation[112–114]. Specifically, belimumab is currently in Phase III clinical trials for investigation of its safety and efficacy in lupus Citation[115]. Of note, after B-cell depletion for autoimmune disease with rituximab, an increase in BLyS levels has been observed, which may allow less stringent negative selection, and therefore the re-emergence of autoreactive B cells Citation[116,117].
Similarly, treatment of alloantigen-naive recipients with rituximab should mandate special consideration of the elevated BLyS levels that may predispose the B-cell repertoire to aberrant negative selection stringency of alloreactive clones Citation[118]. For example, Sollinger et al. found a higher incidence of rejection, especially AMR, in a study of patients treated with induction alemtuzumab and rituximab Citation[119]. However, in the small, randomized trial of rituximab induction therapy, the mean serum levels of APRIL and BLyS were not significantly elevated, although levels were only reported for 14 days after induction Citation[39]. Nonetheless, when patients treated with alemtuzumab were followed-up at least 2 years post-transplant, significantly elevated BLyS levels were found, while patients treated with either calcineurin inhibitors, mycophenolic acid or steroids did not demonstrate increased BLyS levels Citation[120,121]. In addition, in the setting of non-myeloablative conditioning and T-cell induction therapy for kidney and bone marrow transplantation, T-cell tolerance occurred. However, two out of the four patients in this study developed de novo donor-specific HLA antibodies, which were preceded by elevated BLyS levels Citation[10]. Therefore, BLyS neutralization may be an efficacious alternative for B-cell-directed therapy, especially as maintenance therapy.
Also of note, elevated detection of BLyS on CD3+ T-cells has been observed in patients with evidence of allograft rejection at least 5 years after renal transplantation. Interestingly, these patients also had significantly greater levels of anti-HLA class I and II antibodies Citation[122,123]. Overall, these studies highlight how supraphysiologic BLyS – soluble or surface bound – may potentiate an alloreactive B-cell repertoire and may precipitate allograft rejection. BLyS-neutralizing therapy should be investigated following B-cell depletion regimens for its potential to reduce AMR.
Detecting B-cell tolerance & its potential pitfalls
Regardless of the regimen used, the identification of B-cell tolerance should be assessed clinically. For example, methods of B-cell alloimmunity (solid-phase ELISPOT, B-cell flow cytometry on peripheral blood and serum antibodies) should be critically analyzed, such as when B-cell tolerance was identified after infant heart transplantation Citation[36]. However, ELISPOT assays for the visualization of antibody-producing cells should be improved to better detect the polyclonal response that occurs after MHC-mismatched transplantation. Furthermore, B-cell tolerance after transplantation should be tested for by determining the ratio of the absolute number of tolerance-susceptible transitional B cells to follicular B cells Citation[41]. Upon reconstitution of the newly emerging primary repertoire, the amount of circulating and bound BLyS should be determined to optimize therapeutic BLyS neutralization. The amount of donor-specific antibody that is formed after transplantation should be tested regularly to determine the extent of alloreactive clone formation, and hence the potential need for alloantibody-removal treatments (e.g., plasmapheresis) Citation[9]. In addition, tolerance could be monitored with microarrays, to study changes in gene expression Citation[124].
The arguments against our strategy for B-cell tolerance are concerns regarding compromised B-cell immune memory and the dangers of simultaneously targeting both B and T-cells. It should be noted that rituximab-mediated B-cell depletion has shown a minimal impact on the tetanus toxoid memory response in nonhuman primate and human clinical studies Citation[41,125]. Furthermore, belimumab has been generally well tolerated in the clinical studies conducted to date Citation[112,126]. Likewise, the consequences of concurrent B- and T-cell-depletion therapy have been tested in other similar strategies (e.g., alemtuzumab). A 5-year follow-up after induction alemtuzumab therapy demonstrated satisfactory long-term survival and no significant difference from the control groups with respect to infections or serious adverse events, including death Citation[127]. The safety profile of our proposed regimen is likely to be acceptable; however, careful clinical trials for dose safety and efficacy are warranted before widespread application is instituted. For example, during the treatment of acute rejection, alemtuzumab should be used with caution in recipients who underwent induction alemtuzumab or thymoglobulin, as an excess of infection-associated deaths has been observed Citation[128,129]. In summary, B-cell depletion of naive hosts prior to transplantation warrants further investigation in a randomized, controlled clinical trial – ideally using induction T-depletion therapy and an agent that limits BLyS during B-cell repertoire reconstitution Citation[9,101,121].
Antigen-experienced B cells & the loss of B-cell tolerance to alloantigen
The diversity of the pre-immune repertoire is sufficient for basal immunosurveillance Citation[42]. Consequently, once foreign antigen is encountered, if the avidity and extent of B-cell antigen receptor signaling interactions are strong enough and coincident with significant costimulatory signaling, the B-cell undergoes activation Citation[26]. Affinity for antigen is enhanced during the process of somatic hypermutation, in which IgH and IgL variable region genes rapidly undergo base pair substitutions after antigen exposure Citation[130]. B-cell clones with the highest antigen affinity undergo affinity maturation – an antigen-directed B-cell proliferation Citation[131]. During this germinal center reaction, antigen-experienced B cells – liberated from the strict homeostatic constraints of the pre-immune repertoire – diverge into discrete niches, express different BLyS family receptors and become governed by different homeostatic controls Citation[42,43,132,133].
B-cell antigen responses are either TD or T-cell independent (TI) Citation[43,63]. TD responses typically involve follicular B cells and develop in concordance with stimulation from the B-cell antigen receptor and CD40 costimulation Citation[43]. The resulting germinal centers rapidly generate short-lived primary antibody-forming cells. The pre-immune repertoire B cells recruited into this TD response experience selective expansion and preservation, and subsequently become antigen-experienced B cells and long-lived memory B cells Citation[134,135]. Memory B cells have completed class-switch recombination to IgG and occupy a small but heterogeneous fraction of all B cells. These cells persist indefinitely as they demonstrate much slower turnover rates than primary follicular cells Citation[43]. Murine and human studies show memory B cells express increased levels of CD80 and CD95 Citation[136,137]. Human memory B cells also express CD27 Citation[12]. Furthermore, memory B-cell murine investigation has shown increased surface expression of CD73, MHC class II and CD62L Citation[137]. The heightened CD80 and MHC class II expression on memory B cells suggests that antigen presentation to CD4+ T-cells is avid during a recall response Citation[137]. B-cell activation during the TD response, such as during acute cellular rejection, causes the formation of LLPCs – providing a stable supply of antigen-specific antibody, such as donor-specific alloantibody. By contrast, TI responses develop primarily from B1 and marginal zone B cells, which produce short-lived antibody-forming cells Citation[135]. Marginal zone B cells are important for innate and adaptive immunity as they can undergo rapid activation and differentiation into plasma cells Citation[138]. Of note, careful analysis of the primary and secondary response to malaria infection in mice has demonstrated that germinal center B cells and marginal zone B cells expand promptly during a second infection, generating more LLPCs Citation[139]. Overall, the humoral immune system becomes more efficient after repeated infection – a topic that warrants thorough investigation in basic transplantation research.
BLyS family members & the B-cell response to antigen
While pre-immune B cells express both BR3 and TACI, only BR3 is necessary for their survival. All recently activated B cells express one or more of the BLyS family of receptors. TD antigenic stimulus of B cells favors BR3 expression during germinal center reactions, while TACI expression is favored in the short-lived antibody-forming cells of a TI response. For example, A/WySnJ mice demonstrate normal primary IgM responses for both TD and TI antigens. However, these mutant mice exhibit poor secondary humoral responses, low IgG levels and compromised germinal center formation Citation[140–142]. Both BLyS and APRIL are implicated in the primary humoral response (by influencing isotype switching), either directly or indirectly extending survival Citation[63].
The precise role of BLyS family members during B-cell maturation into the long-lived memory B and antibody-forming cells is under active investigation. However, memory B cells do express elevated levels of TACI at least initially, and do not require BLyS or APRIL to survive Citation[143]. Therefore, memory B cells are the only known B2 subset to survive independent of BLyS or APRIL Citation[143]. Short-lived antibody-forming cells express high levels of TACI, and antigen-experienced germinal center B cells express high levels of BR3. The factors influencing LLPC survival are of tremendous importance in transplantation patients who harbor alloantibodies Citation[12,13]. The conversion from the BR3 to the BCMA-directed survival system suggests homeostatic compartmentalization of the LLPC pool from the pre-immune pool. LLPCs require BLyS or APRIL for survival as they express BCMA and reside primarily in the bone marrow Citation[144]. Although a variety of tissues express APRIL, its mRNA expression is very high in human and mouse osteoclasts, implying the importance of this cytokine to LLPC survival Citation[145]. Importantly, in vivo investigations following antigen immunization demonstrated that the absence of either APRIL or BLyS did not limit LLPC survival, but the combined absence of BLyS and APRIL significantly limits LLPC survival Citation[143]. Notably, LLPCs (as well as natural antibody-forming cells and memory B cells) have demonstrated survival after BLyS inhibition Citation[146]. Furthermore, LLPCs are not dependent on memory B cells for survival Citation[147].
B cells & the immune response against an allograft
Historically, there has been controversy regarding the role of B cells and their secreted antibodies during the alloimmune response as passive transfer of serum from sensitized hosts to naive recipients was not sufficient to provoke rejection Citation[148]. Moreover, the fact that B-cell-deficient mice retained their ability to cause allograft rejection lent further support to the notion that B cells do not participate in the pathogenesis of acute alloimmune responses Citation[149,150]. However, further analysis of B-cell-deficient mice revealed that in the absence of B cells there was an increased production of IL-12 by DCs and a marked polarization of the immune response toward a Th1 phenotype Citation[151]. Accordingly, further investigations concluded that three discrete processes characterize the importance of B-lymphocytes during an alloimmune response: humoral (antibody), cellular (B-cell-dependent) and antigen–antibody complex mechanisms Citation[14].
First, alloantibodies secreted by recipient B-lymphocytes bind MHC antigens and vascular endothelium, causing complement activation and subsequent rejection Citation[152]. Acute cellular rejection is mediated. The development of DSAs before rejection prompted Terasaki et al. to conclude that alloantibodies cause chronic rejection and that therapeutic removal of DSAs must occur to prevent allograft failure Citation[9].
Second, B-lymphocytes serve as efficient antigen-presenting cells (APCs; in addition to DCs and macrophages) for activation of alloreactive CD4+ T-cells. For example, cardiac allograft survival was markedly prolonged in mice with deficient in B-cell-mediated antigen presentation Citation[153,154]. The importance of cognate B- and T-cell interactions after transplantation was further corroborated by findings that allograft-specific memory B cells and/or preformed allospecific antibodies facilitated the activation and proliferation of naive alloreactive T-cells, and ultimately rejection, in otherwise tolerant anti-CD154-treated recipients Citation[155]. Although these studies do not determine whether the absence of alloantibodies or the absence of cognate T–B-cell interactions are sufficient to cause prolonged allograft survival, the abrogation of the germinal center reaction, as in the setting of B-cell tolerance, prevents the formation of both, and hence permits prolonged allograft survival Citation[36].
Third, B cells may generate alloantibodies that form soluble alloantigen–alloantibody immune complexes, which can engage complement receptors and Fcγ receptors on APCs. Accordingly, these antigen–antibody complexes will induce APC maturation and subsequent CD4+ and CD8+ alloreactive T-cell activation Citation[156,157].
Challenges of pursuing B-cell tolerance in antigen-experienced patients
Sensitized patients with plasma cells generating high levels of anti-HLA antibodies have low renal transplantation rates, and if they undergo transplantation the rate of allograft loss is unacceptably high Citation[158]. The source of these alloantibodies, LLPCs, are terminally differentiated B cells, which reside within the bone marrow Citation[159]. Continued expression of the following transcription factors is requisite for an ongoing plasma cell phenotype: BLIMP1 and XBP1; conversely, the lack of expression of PAX5, BCL-6 and MTA3 maintains the plasma cell fate Citation[25]. Interestingly, in the murine system CD93 expression is evident on LLPCs and its expression might be important for the maintenance of antibody secretion after TD immunizations Citation[160]. LLPCs are also dependent on the BCMA receptor, as withdrawal of BLyS and APRIL terminates survival Citation[143,144].
Numerous agents have been designed that attempt to dampen the humoral response after sensitization has occurred . Humanized CD20 transgenic mice were developed to analyze the in vivo factors that regulate the mechanisms involved in B-cell immunotherapy Citation[161]. Gong et al. demonstrated that greater than 90% of follicular B cells were depleted after rituximab, leaving only approximately 33% of B220+ splenocytes Citation[162]. This investigation also noted that B-cell populations had a hierarchy of sensitivity to rituximab, starting with the mature follicular pool, followed by the marginal zone and then the germinal center compartments. Importantly, the combination of the anti-hCD20 antibody and BLyS blocking BR3–Fc treatment resulted in the depletion of all splenic B-cell subsets Citation[162]. These results suggest that while anti-CD20 treatment efficiently depletes circulating B cells, cells with reduced exposure to the reticular endothelial system (such as in those in the marginal zone) require other mechanisms for efficient depletion, such as BLyS neutralization Citation[162]. As mentioned previously, the marginal zone and germinal center pools are critical precursors to antibody-forming cells – hence depletion of these pools is needed to intervene therapeutically in the setting of AMR Citation[18]. Consequently, the relatively diminished sensitivity of the marginal and germinal center compartments to rituximab indicates that other treatments are needed to target the plasma cells.
Nonetheless, patients who develop alloantibodies and AMR may be candidates for B-cell depletion therapy with rituximab Citation[11]. Currently, rituximab is the primary B-cell depletion agent available and this literature has been reviewed in Citation[12,163]. Approximately one-third of pediatric and adult acute renal allograft rejection episodes are associated with intragraft CD20+ clusters Citation[19,164–166]. Zarkhin et al. demonstrated in patients with biopsy-proven acute renal allograft rejection (with B-cell infiltrates) that treatment with rituximab could improve calculated creatinine clearance relative to standard-of-care immunosuppression (thymoglobulin and/or pulse steroids) Citation[167]. Interestingly, this treatment caused complete peripheral and intragraft B-cell depletion without a change in the DSA levels Citation[167]. Although rituximab is not known to target LLPCs, it has been proposed to target activated memory cells within the graft Citation[168], and that the lack of continued stimulation of naive B cells would permit DSAs and plasma cells to decline over time Citation[169]. In the setting of transplantation and autoimmunity, rituximab treatment has lead to significant reductions in serum IgM in most studies, while IgG levels are mostly stable Citation[163,167,170]. Consequently, rituximab treatment has been combined with adjunct modalities, such as plasmapheresis, to enhance the effective treatment of AMR Citation[171]. Furthermore, the recent availability of the proteosome inhibitor, bortezomib (which depletes plasma cells) has expanded the scope of intervention strategies to modulate AMR Citation[126,172].
Currently, approximately 30% of patients on the waiting list for renal transplantation have evidence of prior HLA sensitization, and as a result achievement of B-cell tolerance in these patients is impossible with currently instituted protocols Citation[158]. The development of preformed antibodies against HLA occurs as a result of prior transplantation, transfusion and pregnancy. The demand for transplantation in highly sensitized patients continues to climb, as less sensitized patients receive more readily compatible organs – underscoring the need for effective and safe B-cell desensitization protocols. Desensitization strategies have been instituted to improve the likelihood of converting highly sensitized patients with a positive cross-match to a negative cross-match. These protocols employ different combinations of treatment with intravenous Ig (IVIG), rituximab and plasmapheresis with extracorporeal immunoadsorption to remove the DSAs Citation[173,174]. An additional strategy is to perform an extensive search for donors who lack unacceptable HLA specificities reactive with the recipient’s alloantibodies in order to increase the likelihood of a compatible cross-match Citation[175]. Accordingly, although short-term outcomes after desensitization are favorable, medium-to-long-term outcomes fall short of expectations and warrant continued follow-up investigations Citation[175,176]. Interestingly, efforts to induce B-cell tolerance can also be achieved in approximately 70% of patients with hemophilia A who develop inhibitory alloantibodies against plasma-derived and recombinant factor VIII Citation[177]. In these protocols, the use of cyclophosphamide and methotrexate for B-cell depletion, as well as IVIG and plasmapheresis for antibody-cleansing techniques, have been described since the 1970s Citation[178,179].
Several investigations have found utility in rituximab administration for desensitization of highly sensitized patients Citation[18,180–191]. Vo et al. reported that treatment of highly sensitized patients awaiting renal transplantation with rituximab and IVIG lead to a decline in panel reactive antibody from approximately 77 ± 19% to 44 ± 30%, permitting 16 out of 20 of patients to undergo transplantation Citation[158]. The recipients demonstrated mean serum creatinine levels of 1.5 ± 1.1 mg/dl 1 year after transplantation. However, the results of a number of desensitization protocols with rituximab fall short of expectation primarily due to its lack of specificity for LLPCs, cells that do not express CD20 Citation[165]. Furthermore, although rituximab effectively depletes mature peripheral B cells, it has not been shown to consistently diminish the level of preformed alloantibodies Citation[167]. Consequently, other memory B-cell- and plasma cell-selective targets are needed to provide effective desensitization of patients awaiting organ transplantation.
The plasma cell-depleting proteosome inhibitor, bortezomib, in combination with the aforementioned antibody-cleansing techniques, warrants further investigation as a potential alternative to current desensitization protocols Citation[126,172,192]. Furthermore, as was discussed previously, plasma cells require APRIL and BLyS for their survival, among other factors. Interestingly, several autoimmunity studies now report the efficacy of atacicept (formerly TACI-Ig, a fully humanized recombinant fusion protein that binds APRIL and BLyS) for improvement in clinical symptoms and for reduction in serum IgM and rheumatoid factor levels Citation[90,193,194]. In Europe, atacicept is currently in Phase II/III clinical trials for multiple autoimmune disorders and B-cell malignancies Citation[195,196]. In patients with lupus, rheumatoid arthritis and a small cohort of nonhuman primates with circulating donor-specific antibody following rejection of heterotopic heart allografts, atacicept treatment was shown to markedly reduce the Ig levels Citation[197–199]. In summary, novel desensitization protocols targeting the APRIL and BLyS pathways of B-cell homeostasis are warranted in future attempts for the reduction of alloantibodies in sensitized recipients.
Expert commentary & five-year view
The achievement of sustained transplantation tolerance will require a coordinated approach to targeting both branches of the adaptive immune system, that is, T and B-lymphocytes. Currently, the mainstay of clinical immunotherapeutics revolves around T-cell-directed agents. Although these strategies effectively ameliorate the vigor of acute rejection, they fall short of inducing transplantation tolerance. Our main perspective is that, in addition to T-cell-directed immunotherapy, the long-term survival of allografts will require the induction of B-cell tolerance. The alloantibody response by the recipient B-lymphocytes is relevant in two domains as it pertains to organ transplants: first, the B-cell repertoire of an alloantigen-naive recipient confronted with an organ transplant for the first time and, second, an alloantigen experienced recipient with pre-existing alloantibodies and long-lived memory/plasma cells. Based on our current insights into B-cell biology, the naive B-cell repertoire is most amenable to the development of novel and mechanistically based clinical immunotherapeutics. Targeting of the pre-existing recipient memory/plasma cell compartments, on the other hand, will await the development of further insights into the parameters governing the homeostasis of this complex lymphoid subset.
In the case of the alloantigen-naive pre-immune recipient, the primary objective is to prevent the activation and differentiation of alloreactive memory and plasma cells from the resting B-cell repertoire. There is no doubt that interruption of T-cell help to B-lymphocytes using T-cell-directed agents or costimulatory blockade reduces the strength of germinal center reactions, and thereby decreases the potency of the alloantibody response. It is our strong contention here that this tolerogenic effect of T-cell-directed immunotherapy on alloantibody responses could be significantly accentuated by ‘remodeling’ the pre-immune B-cell repertoire itself. A vast body of seminal studies has demonstrated that clonal deletion of immature B cells occurs at discrete checkpoints following exposure to cognate antigen Citation[55–57] and prior to their entry into the mature, pre-immune repertoire Citation[60,62]. The TNF-related cytokine, BLyS, has been identified as the key regulator of these check points and is already a target of clinical tolerance strategies in autoimmunity. We suggest that B-cell depletion using reagents such as anti-CD20 or anti-BLyS agents at the time of transplantation will provide a tolerogenic window for the auto-reconstituting B-cell compartment of the alloantigen-naive transplant recipient to be purged of alloreactive specificities. However, B-cell depletion therapy at the time of induction for the purpose of ‘repertoire remodeling’ is only warranted in conjunction with appropriate T-cell-directed immunotherapeutics to prevent both acute rejection and inhibit the delivery of T-cell help to alloreactive B cells. Overall, the design of clinically relevant approaches grounded in basic mechanistic principles for the purpose of remodeling the primary B-cell repertoire in the setting of organ transplantation should be an important priority over the next few years.
Table 1. Summary of B-cell tolerance-promoting strategies/agents.
Table 2. Potential agents for treating antibody-mediated rejection and/or the presensitized patient.
Key issues
• The long-term survival of organ allografts remains an unfulfilled goal.
• Substantial evidence now implicates B cells as key players in alloimmunity.
• The B-cell alloimmune response, initiated in the germinal center reaction, is typified by the presence of alloantibodies and histopathological evidence of antibody-mediated rejection.
• The TNF-related cytokine, B-lymphocyte stimulator (BLyS), has been identified as the key regulator in naive B-cell homeostasis and it is currently the target of clinical tolerance strategies in autoimmunity.
• B-cell ‘repertoire remodeling’ necessitates B-cell depletion therapy and subsequent repertoire reconstitution in the presence of the allograft and in the absence of excess BLyS, permitting stringent transitional → mature B-cell selection.
• B-cell repertoire remodeling is only warranted in conjunction with appropriate T-cell-directed immunotherapeutics to prevent both acute rejection and to inhibit the delivery of T-cell help to alloreactive B cells.
• The design of logical approaches for the achievement of B-cell tolerance in highly sensitized patients and allograft recipients with antibody-mediated rejection awaits the development of more detailed basic insights into the parameters governing the maintenance of memory/plasma cell subsets.
Financial & competing interests disclosure
Michael Cancro holds a sponsored research agreement with Human Genome Sciences (HGS). Thi-Sau Migone is an employee of HGS who has stock options from the company. This work was supported by the US NIH (grant numbers DK64603 and DK80286 to Hooman Noorchashm) and the Juvenile Diabetes Research Foundation (grant number 5-2008-276 to Ali Naji). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Brennan DC, Daller JA, Lake KD, Cibrik D, Del Castillo D. Rabbit antithymocyte globulin versus basiliximab in renal transplantation. N. Engl. J. Med.355(19), 1967–1977 (2006).
- Pirsch JD, Miller J, Deierhoi MH, Vincenti F, Filo RS. A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression after cadaveric renal transplantation. FK506 Kidney Transplant Study Group. Transplantation63(7), 977–983 (1997).
- Luan FL, Steffick DE, Ojo AO. Steroid-free maintenance immunosuppression in kidney transplantation: is it time to consider it as a standard therapy? Kidney Int. (2009) (Epub ahead of print).
- Deeks ED, Keating GM. Rabbit antithymocyte globulin (thymoglobulin): a review of its use in the prevention and treatment of acute renal allograft rejection. Drugs69(11), 1483–1512 (2009).
- Scherer MN, Banas B, Mantouvalou K et al. Current concepts and perspectives of immunosuppression in organ transplantation. Langenbecks Arch. Surg.392(5), 511–523 (2007).
- Scandling JD, Busque S, Dejbakhsh-Jones S et al. Tolerance and chimerism after renal and hematopoietic-cell transplantation. N. Engl. J. Med.358(4), 362–368 (2008).
- Fudaba Y, Spitzer TR, Shaffer J et al. Myeloma responses and tolerance following combined kidney and nonmyeloablative marrow transplantation: in vivo and in vitro analyses. Am. J. Transplant.6(9), 2121–2133 (2006).
- Fehr T, Sykes M. Clinical experience with mixed chimerism to induce transplantation tolerance. Transpl. Int.21(12), 1118–1135 (2008).
- Terasaki PI, Cai J. Human leukocyte antigen antibodies and chronic rejection: from association to causation. Transplantation86(3), 377–383 (2008).
- Porcheray F, Wong W, Saidman SL et al. B-cell immunity in the context of T-cell tolerance after combined kidney and bone marrow transplantation in humans. Am. J. Transplant.9(9), 2126–2135 (2009).
- Pescovitz MD. B cells: a rational target in alloantibody-mediated solid organ transplantation rejection. Clin. Transplant.20(1), 48–54 (2006).
- Zarkhin V, Li L, Sarwal M. ‘To B or not to B?’ B cells and graft rejection. Transplantation85(12), 1705–1714 (2008).
- Wood KJ. Is B-cell tolerance essential for transplantation tolerance? Transplantation79(Suppl. 3), S40–S42 (2005).
- Vongwiwatana A, Tasanarong A, Hidalgo LG, Halloran PF. The role of B cells and alloantibody in the host response to human organ allografts. Immunol. Rev.196, 197–218 (2003).
- Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr. Opin. Immunol.20(6), 632–638 (2008).
- Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nat. Rev. Immunol.5(10), 807–817 (2005).
- Monroe JG. Molecular mechanisms regulating B-cell responsiveness and tolerance. Transplantation79(Suppl. 3), S12–S13 (2005).
- Becker YT, Samaniego-Picota M, Sollinger HW. The emerging role of rituximab in organ transplantation. Transpl. Int.19(8), 621–628 (2006).
- Sarwal M, Chua MS, Kambham N et al. Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling. N. Engl. J. Med.349(2), 125–138 (2003).
- Colvin RB. Pathology of chronic humoral rejection. Contrib. Nephrol.162, 75–86 (2009).
- Terasaki PI, Ozawa M. Predicting kidney graft failure by HLA antibodies: a prospective trial. Am. J. Transplant.4(3), 438–443 (2004).
- Lachmann N, Terasaki PI, Budde K et al. Anti-human leukocyte antigen and donor-specific antibodies detected by luminex posttransplant serve as biomarkers for chronic rejection of renal allografts. Transplantation87(10), 1505–1513 (2009).
- Wang R, Wang H, Chen J et al. C4d deposition in allograft renal biopsies is an independent risk factor for graft failure. Nephrology (Carlton)14(5), 527–532 (2009).
- Kedainis RL, Koch MJ, Brennan DC, Liapis H. Focal C4d+ in renal allografts is associated with the presence of donor-specific antibodies and decreased allograft survival. Am. J. Transplant.9(4), 812–819 (2009).
- Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat. Rev. Immunol.5(3), 230–242 (2005).
- Schwartzberg PL, Mueller KL, Qi H, Cannons JL. SLAM receptors and SAP influence lymphocyte interactions, development and function. Nat. Rev. Immunol.9(1), 39–46 (2009).
- Rasheed AU, Rahn HP, Sallusto F, Lipp M, Muller G. Follicular B helper T-cell activity is confined to CXCR5hiICOShi CD4 T-cells and is independent of CD57 expression. Eur. J. Immunol.36(7), 1892–1903 (2006).
- McCausland MM, Yusuf I, Tran H, Ono N, Yanagi Y, Crotty S. SAP regulation of follicular helper CD4 T-cell development and humoral immunity is independent of SLAM and Fyn kinase. J. Immunol.178(2), 817–828 (2007).
- Kim CH, Lim HW, Kim JR, Rott L, Hillsamer P, Butcher EC. Unique gene expression program of human germinal center T helper cells. Blood104(7), 1952–1960 (2004).
- Lee PC, Zhu L, Terasaki PI, Everly MJ. HLA-specific antibodies developed in the first year posttransplant are predictive of chronic rejection and renal graft loss. Transplantation88(4), 568–574 (2009).
- Mizutani K, Terasaki P, Rosen A et al. Serial ten-year follow-up of HLA and MICA antibody production prior to kidney graft failure. Am. J. Transplant.5(9), 2265–2272 (2005).
- Sureshkumar KK, Hussain SM, Zimmer BW, Marcus RJ. Emerging role of alemtuzumab in renal and renal-pancreas transplantation. Expert Opin. Biol. Ther.8(10), 1605–1625 (2008).
- Kaufman DB, Leventhal JR, Gallon LG, Parker MA. Alemtuzumab induction and prednisone-free maintenance immunotherapy in simultaneous pancreas–kidney transplantation comparison with rabbit antithymocyte globulin induction – long-term results. Am. J. Transplant.6(2), 331–339 (2006).
- Cai J, Terasaki PI, Bloom DD et al. Correlation between human leukocyte antigen antibody production and serum creatinine in patients receiving sirolimus monotherapy after Campath-1H induction. Transplantation78(6), 919–924 (2004).
- Knechtle SJ, Pirsch JD, H Fechner J Jr et al. Campath-1H induction plus rapamycin monotherapy for renal transplantation: results of a pilot study. Am. J. Transplant.3(6), 722–730 (2003).
- Fan X, Ang A, Pollock-Barziv SM et al. Donor-specific B-cell tolerance after ABO-incompatible infant heart transplantation. Nat. Med.10(11), 1227–1233 (2004).
- West LJ. B-cell tolerance following ABO-incompatible infant heart transplantation. Transplantation81(3), 301–307 (2006).
- Aoyagi T, Yamashita K, Suzuki T et al. A human anti-CD40 monoclonal antibody, 4D11, for kidney transplantation in cynomolgus monkeys: induction and maintenance therapy. Am. J. Transplant.9(8), 1732–1741 (2009).
- Clatworthy MR, Watson CJ, Plotnek G et al. B-cell-depleting induction therapy and acute cellular rejection. N. Engl. J. Med.360(25), 2683–2685 (2009).
- Tarlinton DM, Batista F, Smith KG. The B-cell response to protein antigens in immunity and transplantation. Transplantation85(12), 1698–1704 (2008).
- Liu C, Noorchashm H, Sutter JA et al. B-lymphocyte-directed immunotherapy promotes long-term islet allograft survival in nonhuman primates. Nat. Med.13(11), 1295–1298 (2007).
- Miller JP, Stadanlick JE, Cancro MP. Space, selection, and surveillance: setting boundaries with BLyS. J. Immunol.176(11), 6405–6410 (2006).
- Crowley JE, Scholz JL, Quinn WJ 3rd et al. Homeostatic control of B-lymphocyte subsets. Immunol. Res.42(1–3), 75–83 (2008).
- Osmond DG, Rolink A, Melchers F. Murine B lymphopoiesis: towards a unified model. Immunol. Today19(2), 65–68 (1998).
- Osmond DG, Rico-Vargas S, Valenzona H et al. Apoptosis and macrophage-mediated cell deletion in the regulation of B lymphopoiesis in mouse bone marrow. Immunol. Rev.142, 209–230 (1994).
- Osmond DG, Melchers F, Paige CJ. Pre-B cells in mouse bone marrow: in vitro maturation of peanut agglutinin binding B-lymphocyte precursors separated from bone marrow by fluorescence-activated cell sorting. J. Immunol.133(1), 86–90 (1984).
- Osmond DG. Population dynamics of bone marrow B-lymphocytes. Immunol. Rev.93, 103–124 (1986).
- Melchers F, Rolink A, Grawunder U et al. Positive and negative selection events during B lymphopoiesis. Curr. Opin. Immunol.7(2), 214–227 (1995).
- Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K. Resolution and characterization of pro-B and pre-pro-B-cell stages in normal mouse bone marrow. J. Exp. Med.173(5), 1213–1225 (1991).
- Hardy RR, Li YS, Allman D, Asano M, Gui M, Hayakawa K. B-cell commitment, development and selection. Immunol. Rev.175, 23–32 (2000).
- Li Y, Ma L, Shen J, Chong AS. Peripheral deletion of mature alloreactive B cells induced by costimulation blockade. Proc. Natl Acad. Sci. USA104(29), 12093–12098 (2007).
- Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature435(7042), 590–597 (2005).
- Goodnow CC. Nossal and Pike 1975: a turning point in the effort to define self-tolerance mechanisms. J. Immunol.179(9), 5617–5618 (2007).
- Nossal GJ, Pike BL. Evidence for the clonal abortion theory of B-lymphocyte tolerance. J. Exp. Med.141(4), 904–917 (1975).
- Norvell A, Mandik L, Monroe JG. Engagement of the antigen-receptor on immature murine B-lymphocytes results in death by apoptosis. J. Immunol.154(9), 4404–4413 (1995).
- Nemazee DA, Burki K. Clonal deletion of B-lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature337(6207), 562–566 (1989).
- Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC. Elimination from peripheral lymphoid tissues of self-reactive B-lymphocytes recognizing membrane-bound antigens. Nature353(6346), 765–769 (1991).
- Nemazee D, Buerki K. Clonal deletion of autoreactive B-lymphocytes in bone marrow chimeras. Proc. Natl Acad. Sci. USA86(20), 8039–8043 (1989).
- Russell DM, Dembic Z, Morahan G, Miller JF, Burki K, Nemazee D. Peripheral deletion of self-reactive B cells. Nature354(6351), 308–311 (1991).
- Cyster JG, Hartley SB, Goodnow CC. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature371(6496), 389–395 (1994).
- Shokat KM, Goodnow CC. Antigen-induced B-cell death and elimination during germinal-centre immune responses. Nature375(6529), 334–338 (1995).
- Cyster JG, Goodnow CC. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B-cell fate. Immunity3(6), 691–701 (1995).
- Treml JF, Hao Y, Stadanlick JE, Cancro MP. The BLyS family: toward a molecular understanding of B-cell homeostasis. Cell Biochem. Biophys53(1), 1–16 (2009).
- Miosge LA, Goodnow CC. Genes, pathways and checkpoints in lymphocyte development and homeostasis. Immunol. Cell Biol.83(4), 318–335 (2005).
- Rolink AG, Schaniel C, Andersson J, Melchers F. Selection events operating at various stages in B-cell development. Curr. Opin. Immunol.13(2), 202–207 (2001).
- Louzoun Y, Friedman T, Luning Prak E, Litwin S, Weigert M. Analysis of B-cell receptor production and rearrangement. Part I. Light chain rearrangement. Semin. Immunol.14(3), 169–190; discussion 221–122 (2002).
- Liu Y, Li L, Kumar KR et al. Lupus susceptibility genes may breach tolerance to DNA by impairing receptor editing of nuclear antigen-reactive B cells. J. Immunol.179(2), 1340–1352 (2007).
- Verkoczy LK, Martensson AS, Nemazee D. The scope of receptor editing and its association with autoimmunity. Curr. Opin. Immunol.16(6), 808–814 (2004).
- Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J. Exp. Med.177(4), 999–1008 (1993).
- Kouskoff V, Lacaud G, Pape K, Retter M, Nemazee D. B-cell receptor expression level determines the fate of developing B-lymphocytes: receptor editing versus selection. Proc. Natl Acad. Sci. USA97(13), 7435–7439 (2000).
- Stadanlick JE, Kaileh M, Karnell FG et al. Tonic B-cell antigen receptor signals supply an NF-κB substrate for prosurvival BLyS signaling. Nat. Immunol.9(12), 1379–1387 (2008).
- Goodnow CC, Crosbie J, Adelstein S et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature334(6184), 676–682 (1988).
- Erikson J, Radic MZ, Camper SA, Hardy RR, Carmack C, Weigert M. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature349(6307), 331–334 (1991).
- Treml LS, Quinn WJ 3rd, Treml JF, Scholz JL, Cancro MP. Manipulating B-cell homeostasis: a key component in the advancement of targeted strategies. Arch. Immunol. Ther. Exp. (Warsz)56(3), 153–164 (2008).
- Yan M, Brady JR, Chan B et al. Identification of a novel receptor for B-lymphocyte stimulator that is mutated in a mouse strain with severe B-cell deficiency. Curr. Biol.11(19), 1547–1552 (2001).
- Smith SH, Cancro MP. Cutting edge: B-cell receptor signals regulate BLyS receptor levels in mature B cells and their immediate progenitors. J. Immunol.170(12), 5820–5823 (2003).
- Mackay F, Woodcock SA, Lawton P et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med.190(11), 1697–1710 (1999).
- Batten M, Groom J, Cachero TG et al. BAFF mediates survival of peripheral immature B-lymphocytes. J. Exp. Med.192(10), 1453–1466 (2000).
- Moore PA, Belvedere O, Orr A et al. BLyS: member of the tumor necrosis factor family and B-lymphocyte stimulator. Science285(5425), 260–263 (1999).
- Schneider P, MacKay F, Steiner V et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B-cell growth. J. Exp. Med.189(11), 1747–1756 (1999).
- Warnatz K, Salzer U, Rizzi M et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc. Natl Acad. Sci. USA106(33), 13945–13950 (2009).
- Kalled SL. The role of BAFF in immune function and implications for autoimmunity. Immunol. Rev.204, 43–54 (2005).
- Hsu BL, Harless SM, Lindsley RC, Hilbert DM, Cancro MP. Cutting edge: BLyS enables survival of transitional and mature B cells through distinct mediators. J. Immunol.168(12), 5993–5996 (2002).
- Enzler T, Bonizzi G, Silverman GJ et al. Alternative and classical NF-κ B signaling retain autoreactive B cells in the splenic marginal zone and result in lupus-like disease. Immunity25(3), 403–415 (2006).
- Hatada EN, Do RK, Orlofsky A et al. NF-κ B1 p50 is required for BLyS attenuation of apoptosis but dispensable for processing of NF-κ B2 p100 to p52 in quiescent mature B cells. J. Immunol.171(2), 761–768 (2003).
- Woodland RT, Fox CJ, Schmidt MR et al. Multiple signaling pathways promote B-lymphocyte stimulator dependent B-cell growth and survival. Blood111(2), 750–760 (2008).
- Day ES, Cachero TG, Qian F et al. Selectivity of BAFF/BLyS and APRIL for binding to the TNF family receptors BAFFR/BR3 and BCMA. Biochemistry44(6), 1919–1931 (2005).
- Cao M, Chen L, Shan XX, Zhang SQ. Immunological effects of refolded human soluble BAFF synthesized in Escherichia coli on murine B-lymphocytes in vitro and in vivo. Jpn J. Physiol.55(4), 221–227 (2005).
- Hymowitz SG, Patel DR, Wallweber HJ et al. Structures of APRIL-receptor complexes: like BCMA, TACI employs only a single cysteine-rich domain for high affinity ligand binding. J. Biol. Chem.280(8), 7218–7227 (2005).
- Dillon SR, Gross JA, Ansell SM, Novak AJ. An APRIL to remember: novel TNF ligands as therapeutic targets. Nat. Rev. Drug Discov.5(3), 235–246 (2006).
- Harless SM, Lentz VM, Sah AP et al. Competition for BLyS-mediated signaling through Bcmd/BR3 regulates peripheral B-lymphocyte numbers. Curr. Biol.11(24), 1986–1989 (2001).
- Shulga-Morskaya S, Dobles M, Walsh ME et al. B-cell-activating factor belonging to the TNF family acts through separate receptors to support B-cell survival and T-cell-independent antibody formation. J. Immunol.173(4), 2331–2341 (2004).
- Grech AP, Amesbury M, Chan T, Gardam S, Basten A, Brink R. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-κB activation in mature B cells. Immunity21(5), 629–642 (2004).
- Chung JB, Silverman M, Monroe JG. Transitional B cells: step by step towards immune competence. Trends Immunol.24(6), 343–349 (2003).
- Yu M, Parsons RF, Rostami Set al.In vivo BLyS/BAFF neutralization induces donor specific transplantation tolerance to murine islet allografts. Am. J. Transplant.9(Suppl. 2), 728 (2009).
- Morecki S, Leshem B, Eid A, Slavin S. Alloantigen persistence in induction and maintenance of transplantation tolerance. J. Exp. Med.165(6), 1468–1480 (1987).
- Hamano K, Rawsthorne MA, Bushell AR, Morris PJ, Wood KJ. Evidence that the continued presence of the organ graft and not peripheral donor microchimerism is essential for maintenance of tolerance to alloantigen in vivo in anti-CD4 treated recipients. Transplantation62(6), 856–860 (1996).
- Lang J, Nemazee D. B-cell clonal elimination induced by membrane-bound self-antigen may require repeated antigen encounter or cell competition. Eur. J. Immunol.30(2), 689–696 (2000).
- Sykes M, Shimizu I, Kawahara T. Mixed hematopoietic chimerism for the simultaneous induction of T and B-cell tolerance. Transplantation79(Suppl. 3), S28–S29 (2005).
- Yang YG, deGoma E, Ohdan H et al. Tolerization of anti-Galα1–3Gal natural antibody-forming B cells by induction of mixed chimerism. J. Exp. Med.187(8), 1335–1342 (1998).
- Terasaki PI, Ozawa M, Castro R. Four-year follow-up of a prospective trial of HLA and MICA antibodies on kidney graft survival. Am. J. Transplant.7(2), 408–415 (2007).
- Yanaba K, Bouaziz JD, Matsushita T, Tsubata T, Tedder TF. The development and function of regulatory B cells expressing IL-10 (B10 cells) requires antigen receptor diversity and TLR signals. J. Immunol.182(12), 7459–7472 (2009).
- Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Invest.118(10), 3420–3430 (2008).
- Moore KW, O’Garra A, de Waal Malefyt R, Vieira P, Mosmann TR. Interleukin-10. Annu. Rev. Immunol.11, 165–190 (1993).
- Gross JA, Dillon SR, Mudri S et al. TACI-Ig neutralizes molecules critical for B-cell development and autoimmune disease. Impaired B-cell maturation in mice lacking BLyS. Immunity15(2), 289–302 (2001).
- Thien M, Phan TG, Gardam S et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity20(6), 785–798 (2004).
- Brink R. Regulation of B-cell self-tolerance by BAFF. Semin. Immunol.18(5), 276–283 (2006).
- Quinn WJ 3rd, Noorchashm N, Crowley JE et al. Cutting edge: impaired transitional B-cell production and selection in the nonobese diabetic mouse. J. Immunol.176(12), 7159–7164 (2006).
- Zekavat G, Rostami SY, Badkerhanian Aet al.In vivo BLyS/BAFF neutralization ameliorates islet-directed autoimmunity in nonobese diabetic mice. J. Immunol.181(11), 8133–8144 (2008).
- Kahn P, Ramanujam M, Bethunaickan R et al. Prevention of murine antiphospholipid syndrome by BAFF blockade. Arthritis Rheum.58(9), 2824–2834 (2008).
- Marino E, Villanueva J, Walters S, Liuwantara D, Mackay F, Grey ST. CD4+CD25+ T-cells control autoimmunity in the absence of B cells. Diabetes58(7), 1568–1577 (2009).
- Ding C. Belimumab, an anti-BLyS human monoclonal antibody for potential treatment of inflammatory autoimmune diseases. Expert Opin. Biol. Ther.8(11), 1805–1814 (2008).
- Furie R, Stohl W, Ginzler EM et al. Biologic activity and safety of belimumab, a neutralizing anti-B-lymphocyte stimulator (BLyS) monoclonal antibody: a Phase I trial in patients with systemic lupus erythematosus. Arthritis Res. Ther.10(5), R109 (2008).
- Dorner T. Crossroads of B-cell activation in autoimmunity: rationale of targeting B cells. J. Rheumatol. Suppl.77, 3–11 (2006).
- Ding C, Jones G. Belimumab Human Genome Sciences/Cambridge Antibody Technology/GlaxoSmithKline. Curr. Opin. Investig. Drugs7(5), 464–472 (2006).
- Lavie F, Miceli-Richard C, Ittah M, Sellam J, Gottenberg JE, Mariette X. Increase of B-cell-activating factor of the TNF family (BAFF) after rituximab treatment: insights into a new regulating system of BAFF production. Ann. Rheum. Dis.66(5), 700–703 (2007).
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B-lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B-cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum.54(3), 723–732 (2006).
- Pers JO, Devauchelle V, Daridon C et al. BAFF-modulated repopulation of B lymphocytes in the blood and salivary glands of rituximab-treated patients with Sjögren’s syndrome. Arthritis Rheum.56(5), 1464–1477 (2007).
- Sundberg A, Samaniego M, Pirsch J et al. A pilot study of campath-1H and rituximab induction therapy combined with Cellcept to allow for a calcineurin inhibitor-free regimen after renal transplantation. Am. J. Transplant.9(Suppl. 2), 258 (2009).
- Bloom DD, Pauly K, Chang Z et al. BAFF dysregulation in renal transplant patients treated with campath-1H. Am. J. Transplant.9(Suppl. 2), 226 (2009).
- Bloom D, Chang Z, Pauly K et al. BAFF is increased in renal transplant patients following treatment with alemtuzumab. Am. J. Transplant.9(8), 1835–1845 (2009).
- Xu H, He X, Liu Q et al. The abnormal high expression of B-cell activating factor belonging to TNF superfamily (BAFF) and its potential role in kidney transplant recipients. Cell Mol. Immunol.5(6), 465–470 (2008).
- Xu H, He X, Liu Q et al. Abnormal high expression of B-cell activating factor belonging to the TNF superfamily (BAFF) associated with long-term outcome in kidney transplant recipients. Transplant Proc.41(5), 1552–1556 (2009).
- Zarkhin V, Sarwal MM. Microarrays: monitoring for transplant tolerance and mechanistic insights. Clin. Lab. Med.28(3), 385–410, vi (2008).
- Cambridge G, Leandro MJ, Teodorescu M et al. B-cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum.54(11), 3612–3622 (2006).
- Everly MJ, Everly JJ, Susskind B et al. Bortezomib provides effective therapy for antibody- and cell-mediated acute rejection. Transplantation86(12), 1754–1761 (2008).
- Watson CJ, Bradley JA, Friend PJ et al. Alemtuzumab (campath 1H) induction therapy in cadaveric kidney transplantation – efficacy and safety at five years. Am. J. Transplant.5(6), 1347–1353 (2005).
- Basu A, Ramkumar M, Tan HP et al. Reversal of acute cellular rejection after renal transplantation with campath-1H. Transplant Proc.37(2), 923–926 (2005).
- Clatworthy MR, Friend PJ, Calne RY et al. Alemtuzumab (campath-1H) for the treatment of acute rejection in kidney transplant recipients: long-term follow-up. Transplantation87(7), 1092–1095 (2009).
- Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem.76, 1–22 (2007).
- Rajewsky K. Clonal selection and learning in the antibody system. Nature381(6585), 751–758 (1996).
- Tarlinton D, Radbruch A, Hiepe F, Dorner T. Plasma cell differentiation and survival. Curr. Opin. Immunol.20(2), 162–169 (2008).
- Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B-cell segregation between the outer and centre follicle. Nature460(7259), 1122–1126 (2009).
- Yamauchi T, Ueki K, Tobe K et al. Tyrosine phosphorylation of the EGF receptor by the kinase Jak2 is induced by growth hormone. Nature390(6655), 91–96 (1997).
- Zubler RH. Naive and memory B cells in T-cell-dependent and T-independent responses. Springer Semin. Immunopathol.23(4), 405–419 (2001).
- Bar-Or A, Oliveira EM, Anderson DE et al. Immunological memory: contribution of memory B cells expressing costimulatory molecules in the resting state. J. Immunol.167(10), 5669–5677 (2001).
- Anderson SM, Tomayko MM, Ahuja A, Haberman AM, Shlomchik MJ. New markers for murine memory B cells that define mutated and unmutated subsets. J. Exp. Med.204(9), 2103–2114 (2007).
- Lopes-Carvalho T, Kearney JF. Marginal zone B-cell physiology and disease. Curr. Dir. Autoimmun.8, 91–123 (2005).
- Stephens R, Ndungu FM, Langhorne J. Germinal centre and marginal zone B cells expand quickly in a second Plasmodium chabaudi malaria infection producing mature plasma cells. Parasite Immunol.31(1), 20–31 (2009).
- Miller DJ, Hayes CE. Phenotypic and genetic characterization of a unique B-lymphocyte deficiency in strain A/WySnJ mice. Eur. J. Immunol.21(5), 1123–1130 (1991).
- Miller DJ, Hanson KD, Carman JA, Hayes CE. A single autosomal gene defect severely limits IgG but not IgM responses in B-lymphocyte-deficient A/WySnJ mice. Eur. J. Immunol.22(2), 373–379 (1992).
- Rahman ZS, Manser T. B cells expressing Bcl-2 and a signaling-impaired BAFF-specific receptor fail to mature and are deficient in the formation of lymphoid follicles and germinal centers. J. Immunol.173(10), 6179–6188 (2004).
- Benson MJ, Dillon SR, Castigli E et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J. Immunol.180(6), 3655–3659 (2008).
- O’Connor BP, Raman VS, Erickson LD et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med.199(1), 91–98 (2004).
- Moreaux J, Cremer FW, Reme T et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood106(3), 1021–1030 (2005).
- Scholz JL, Crowley JE, Tomayko MM et al. BLyS inhibition eliminates primary B cells but leaves natural and acquired humoral immunity intact. Proc. Natl Acad. Sci. USA105(40), 15517–15522 (2008).
- Ahuja A, Anderson SM, Khalil A, Shlomchik MJ. Maintenance of the plasma cell pool is independent of memory B cells. Proc. Natl Acad. Sci. USA105(12), 4802–4807 (2008).
- Mitchison NA. Studies on the immunological response to foreign tumor transplants in the mouse. I. The role of lymph node cells in conferring immunity by adoptive transfer. J. Exp. Med.102(2), 157–177 (1955).
- Di Rosa F, Matzinger P. Long-lasting CD8 T-cell memory in the absence of CD4 T-cells or B cells. J. Exp. Med.183(5), 2153–2163 (1996).
- Brandle D, Joergensen J, Zenke G, Burki K, Hof RP. Contribution of donor-specific antibodies to acute allograft rejection: evidence from B-cell-deficient mice. Transplantation65(11), 1489–1493 (1998).
- Moulin V, Andris F, Thielemans K, Maliszewski C, Urbain J, Moser M. B-lymphocytes regulate dendritic cell (DC) function in vivo: increased interleukin 12 production by DCs from B-cell-deficient mice results in T helper cell type 1 deviation. J. Exp. Med.192(4), 475–482 (2000).
- Wasowska BA, Qian Z, Cangello DL et al. Passive transfer of alloantibodies restores acute cardiac rejection in IgKO mice. Transplantation71(6), 727–736 (2001).
- Noorchashm H, Reed AJ, Rostami SY et al. B-cell-mediated antigen presentation is required for the pathogenesis of acute cardiac allograft rejection. J. Immunol.177(11), 7715–7722 (2006).
- Ng YH, Li Q, Marlowe A, Hoffman R, Chalasani G. Antigen presentation by B cells promotes heart allograft rejection and development of alloreactive memory T-cells. Am. J. Transplant.8(Suppl. 2), 177–336 (2008).
- Burns AM, Ma L, Li Y et al. Memory alloreactive B cells and alloantibodies prevent anti-CD154-mediated allograft acceptance. J. Immunol.182(3), 1314–1324 (2009).
- Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J. Clin. Invest.115(10), 2914–2923 (2005).
- Yada A, Ebihara S, Matsumura K et al. Accelerated antigen presentation and elicitation of humoral response in vivo by FcgRIIB- and FcgRI/III-mediated immune complex uptake. Cell Immunol.225(1), 21–32 (2003).
- Vo AA, Lukovsky M, Toyoda M et al. Rituximab and intravenous immune globulin for desensitization during renal transplantation. N. Engl. J. Med.359(3), 242–251 (2008).
- Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science298(5601), 2199–2202 (2002).
- Chevrier S, Genton C, Kallies A et al. CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proc. Natl Acad. Sci. USA106(10), 3895–3900 (2009).
- Hu CY, Rodriguez-Pinto D, Du W et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J. Clin. Invest.117(12), 3857–3867 (2007).
- Gong Q, Ou Q, Ye S et al. Importance of cellular microenvironment and circulatory dynamics in B-cell immunotherapy. J. Immunol.174(2), 817–826 (2005).
- Cornec D, Avouac J, Youinou P, Saraux A. Critical analysis of rituximab-induced serological changes in connective tissue diseases. Autoimmun. Rev.8(6), 515–519 (2009).
- Tsai EW, Rianthavorn P, Gjertson DW, Wallace WD, Reed EF, Ettenger RB. CD20+ lymphocytes in renal allografts are associated with poor graft survival in pediatric patients. Transplantation82(12), 1769–1773 (2006).
- Hippen BE, DeMattos A, Cook WJ, Kew CE 2nd, Gaston RS. Association of CD20+ infiltrates with poorer clinical outcomes in acute cellular rejection of renal allografts. Am. J. Transplant.5(9), 2248–2252 (2005).
- Martins HL, Silva C, Martini D, Noronha IL. Detection of B-lymphocytes (CD20+) in renal allograft biopsy specimens. Transplant Proc.39(2), 432–434 (2007).
- Zarkhin V, Li L, Kambham N, Sigdel T, Salvatierra O, Sarwal MM. A randomized, prospective trial of rituximab for acute rejection in pediatric renal transplantation. Am. J. Transplant.8(12), 2607–2617 (2008).
- Zarkhin V, Kambham N, Li L et al. Characterization of intra-graft B cells during renal allograft rejection. Kidney Int.74(5), 664–673 (2008).
- Pescovitz MD. The use of rituximab, anti-CD20 monoclonal antibody, in pediatric transplantation. Pediatr. Transplant.8(1), 9–21 (2004).
- Vallerskog T, Gunnarsson I, Widhe M et al. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin. Immunol.122(1), 62–74 (2007).
- Faguer S, Kamar N, Guilbeaud-Frugier C et al. Rituximab therapy for acute humoral rejection after kidney transplantation. Transplantation83(9), 1277–1280 (2007).
- Trivedi HL, Terasaki PI, Feroz A et al. Abrogation of anti-HLA antibodies via proteasome inhibition. Transplantation87(10), 1555–1561 (2009).
- Doxiadis, II, Duquesnoy RJ, Claas FH. Extending options for highly sensitized patients to receive a suitable kidney graft. Curr. Opin. Immunol.17(5), 536–540 (2005).
- Beimler JH, Susal C, Zeier M. Desensitization strategies enabling successful renal transplantation in highly sensitized patients. Clin. Transplant.20(Suppl. 17), 7–12 (2006).
- Doxiadis II, Claas FH. Transplantation of highly sensitized patients via the acceptable mismatch program or desensitization? We need both. Curr. Opin. Organ Transplant14(4), 410–413 (2009).
- Haririan A, Nogueira J, Kukuruga D et al. Positive cross-match living donor kidney transplantation: longer-term outcomes. Am. J. Transplant.9(3), 536–542 (2009).
- DiMichele DM, Kroner BL. The North American Immune Tolerance Registry: practices, outcomes, outcome predictors. Thromb. Haemost.87(1), 52–57 (2002).
- Kempton CL, White GC 2nd. How we treat a hemophilia A patient with a factor VIII inhibitor. Blood113(1), 11–17 (2009).
- Nilsson IM, Berntorp E, Zettervall O. Induction of immune tolerance in patients with hemophilia and antibodies to factor VIII by combined treatment with intravenous IgG, cyclophosphamide, and factor VIII. N. Engl. J. Med.318(15), 947–950 (1988).
- Gloor JM, DeGoey SR, Pineda AA et al. Overcoming a positive crossmatch in living-donor kidney transplantation. Am. J. Transplant.3(8), 1017–1023 (2003).
- Beimler JH, Morath C, Schmidt J et al. Successful deceased-donor kidney transplantation in crossmatch-positive patients with peritransplant plasma exchange and rituximab. Transplantation87(5), 668–671 (2009).
- Tanriover B, Wright SE, Foster SV et al. High-dose intravenous immunoglobulin and rituximab treatment for antibody-mediated rejection after kidney transplantation: a cost analysis. Transplant Proc.40(10), 3393–3396 (2008).
- Ravichandran P, Natrajan T, Jaganathan R. Combination treatment of low dose anti-thymocyte globulin (ATG), rituximab and high dose sirolimus as induction agents in immune-conditioned recipients. Int. Immunopharmacol.6(13–14), 1973–1976 (2006).
- Tanabe K, Ishida H, Shimizu T, Omoto K, Shirakawa H, Tokumoto T. Evaluation of two different preconditioning regimens for ABO-incompatible living kidney donor transplantation. A comparison of splenectomy vs. rituximab-treated non-splenectomy preconditioning regimens. Contrib. Nephrol.162, 61–74 (2009).
- Genberg H, Kumlien G, Wennberg L, Berg U, Tyden G. ABO-incompatible kidney transplantation using antigen-specific immunoadsorption and rituximab: a 3-year follow-up. Transplantation85(12), 1745–1754 (2008).
- Yoon HE, Hyoung BJ, Hwang HS et al. Successful renal transplantation with desensitization in highly sensitized patients: a single center experience. J. Korean Med. Sci.24(Suppl.), S148–S155 (2009).
- Munoz AS, Rioveros AA, Cabanayan-Casasola CB, Danguilan RA, Ona ET. Rituximab in highly sensitized kidney transplant recipients. Transplant Proc.40(7), 2218–2221 (2008).
- Amante AJ, Ejercito R. Management of highly sensitized patients: Capitol Medical Center experience. Transplant Proc.40(7), 2274–2280 (2008).
- Ignjatovic L, Kovacevic Z, Jovanovic D et al. Our first experiences in applying an original method for removal of ABO-isoagglutinins in ABO-incompatible kidney recipients. Vojnosanit Pregl.66(2), 117–122 (2009).
- Reinsmoen NL, Lai CH, Vo A et al. Acceptable donor-specific antibody levels allowing for successful deceased and living donor kidney transplantation after desensitization therapy. Transplantation86(6), 820–825 (2008).
- Vieira CA, Agarwal A, Book BK et al. Rituximab for reduction of anti-HLA antibodies in patients awaiting renal transplantation: 1. Safety, pharmacodynamics, and pharmacokinetics. Transplantation77(4), 542–548 (2004).
- Perry DK, Burns JM, Pollinger HS et al. Proteasome inhibition causes apoptosis of normal human plasma cells preventing alloantibody production. Am. J. Transplant.9(1), 201–209 (2009).
- Tak PP, Thurlings RM, Rossier C et al. Atacicept in patients with rheumatoid arthritis: results of a multicenter, Phase Ib, double-blind, placebo-controlled, dose–escalating, single- and repeated-dose study. Arthritis Rheum.58(1), 61–72 (2008).
- Gross JA, Johnston J, Mudri S et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature404(6781), 995–999 (2000).
- Gatto B. Atacicept, a homodimeric fusion protein for the potential treatment of diseases triggered by plasma cells. Curr. Opin. Investig. Drugs9(11), 1216–1227 (2008).
- Ponce R. Preclinical support for combination therapy in the treatment of autoimmunity with atacicept. Toxicol. Pathol.37(1), 89–99 (2009).
- Bracewell C, Isaacs JD, Emery P, Ng WF. Atacicept, a novel B-cell-targeting biological therapy for the treatment of rheumatoid arthritis. Expert Opin. Biol. Ther.9(7), 909–919 (2009).
- Dall’Era M, Chakravarty E, Wallace D et al. Reduced B-lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, Phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum.56(12), 4142–4150 (2007).
- Boskovic S, Smith NR, Kawai T et al. Inhibitory effects of atacicept (TACI-Ig) on circulating antibodies, B cells and plasma cells in allosensitized cynomolgous monkeys. Am. J. Transplant.8(Suppl. 2), 177–336 (2008).