Abstract
The research reviewed in this article provides examples of autoantibody-mediated receptor activation that likely contributes to disease. The classic example is Graves’ hyperthyroidism, in which autoantibodies activate the thyroid-stimulating hormone receptor resulting in overproduction of thyroid hormones. Other compelling examples come from the cardiovascular literature and include agonistic autoantibodies targeting the cardiac β1-adrenergic receptor, which are associated with dilated cardiomyopathy. Autoantibodies capable of activating α1-adrenergic receptors are associated with refractory hypertension and cardiomyopathy. A prominent example is preeclampsia, a hypertensive disease of pregnancy, characterized by the presence of autoantibodies that activate the major angiotensin receptor, AT1. AT1 receptor-activating autoantibodies are also observed in kidney transplant recipients suffering from severe vascular rejection and malignant hypertension. AT1 receptor-activating autoantibodies and antibodies that activate the endothelin-1 receptor, ETA, are prevalent in individuals diagnosed with systemic sclerosis. Thus, the presence of agonistic autoantibodies directed to G protein-coupled receptors has been observed in numerous cardiovascular disease states. Rapidly emerging evidence indicates that receptor-activating autoantibodies contribute to disease, and that efforts to detect and remove these pathogenic autoantibodies or block their actions will provide promising therapeutic possibilities.
Thyroid-stimulating hormone receptor (TSHR) has an unusually large extracellular domain consisting of nine leucine-rich repeats that form a horseshoe-like structure that binds thyroid-stimulating hormone. Upon proteolytic cleavage resulting in the loss of approximately 50 amino acid residues (˜316–366), TSHR forms a two-subunit structure (α and β) linked by disulfide bonds.
Adapted with permission from Citation[105].
![Figure 1. Thyroid-stimulating hormone receptor subunit structure with bound ligand.Thyroid-stimulating hormone receptor (TSHR) has an unusually large extracellular domain consisting of nine leucine-rich repeats that form a horseshoe-like structure that binds thyroid-stimulating hormone. Upon proteolytic cleavage resulting in the loss of approximately 50 amino acid residues (˜316–366), TSHR forms a two-subunit structure (α and β) linked by disulfide bonds.Adapted with permission from Citation[105].](/cms/asset/ded33cb5-9e09-4498-8841-09c1a7b0d40a/ierm_a_11212220_f0001_b.jpg)
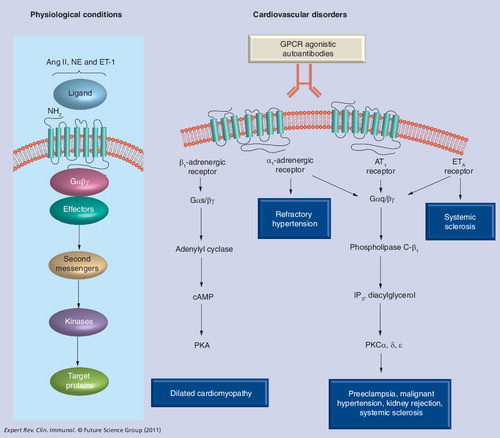
AT1, ETA, α1- and β1-adrenergic receptors are important cardiovascular GPCRs that regulate cardiac function and blood pressure via activation by their respective ligands (Ang II, ET-1 and norepinephrine, respectively). In recent years, GPCR-AAs have been associated with multiple cardiovascular disorders and autoantibody titers correlated with disease severity. Additional evidence reviewed here supports a contributing role for these autoantibodies in disease pathogenesis. These findings highlight these pathogenic autoantibodies as potentially important presymptomatic biomarkers and therapeutic targets in the medical management of these diseases.
AA: Activating autoantibody; GPCR: G protein-coupled receptor.
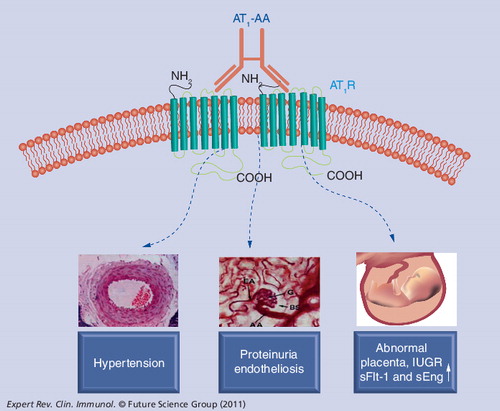
Recent evidence indicates that AT1-AA injection into pregnant rodents (mice or rats) results in multiple feature of preeclampsia, including IUGR. This experimental model of autoantibody-induced preeclampsia and IUGR in pregnant rodents provides important experimental opportunities to test the ability of various strategies to prevent or reverse autoantibody-induced clinical features associated with preeclampsia.
AT1-AA: Angiotensin receptor agonistic autoantibody; AT1R: Angiotensin II receptor; IUGR: Intrauterine growth retardation.
The immune system has sophisticated effector mechanisms to identify foreign antigens and eliminate them from our bodies. Thus, a critical feature of the immune system is the ability to discriminate between self and nonself. When there is a failure in the immunological control mechanisms that maintain self-tolerance, the adaptive immune system can be directed at a self antigen. In most cases the autoantigen continues to be produced, and thus cannot be eliminated by the immune response. In these cases, the adaptive immune response becomes chronic, resulting in disruption of normal physiological functions. Autoimmune diseases vary considerably in the symptoms they cause and in the tissues and organ systems they affect. Autoimmune diseases are relatively common (5% of the US population) and include well known diseases such as Type 1 diabetes, multiple sclerosis, systemic lupus erythematosus, Graves’ disease, myasthenia gravis and rheumatoid arthritis.
Autoimmune diseases can be caused by autoantibodies that disturb normal physiological functions or by cytotoxic T cells that cause targeted cell death. A defining feature of autoimmune disease is the presence of autoantibodies or autoreactive T cells specific for autoantigens present on target cells. A number of autoimmune diseases result from the presence of autoantibodies that bind to G protein-coupled receptors (GPCRs) on the surface of cells. Some diseases, such as Graves’ disease (see later) are characterized by the presence of autoantibodies that activate the target GPCRs. Other diseases (e.g., myasthenia gravis) are characterized by the presence of autoantibodies that bind to receptors, but do not activate. In the latter case, the receptor-bound autoantibodies can recruit complement and lead to cellular destruction. Research reviewed here deals with autoimmune diseases in which the disease pathology results from the presence of autoantibodies that activate cell surface receptors of the GPCR family. In several cases the ability of these autoantibodies to cause disease has been demonstrated by adoptive transfer experiments showing that the transfer of antibody from an affected individual to other humans or laboratory animals is sufficient to cause disease. More recent evidence shows that the removal of pathogenic antibodies by plasmaphoresis or immunoabsorption provides therapeutic benefit.
Graves’ disease
Background
Graves’ disease is a relatively common autoimmune disease characterized by thyroid diffuse enlargement, palpitation, muscle fatigue, tremor and anxiety, collectively termed ‘hyperthyroidism’ Citation[1,2]. The condition affects approximately 1 in 50 women and results from the presence of autoantibodies that activate the thyroid stimulating hormone receptor (TSHR), also called thyrotropin receptor Citation[3]. Graves’ disease was the first autoimmune disease that was characterized by the presence of receptor-activating autoantibodies. Initial evidence was provided by the pioneering studies by Adams and colleagues who described an activity termed ‘long-acting thyroid stimulator’ (LATS) in the serum immunoglobulin fraction of patients with hyperthyroid Graves’ disease. Infusion of LATS into healthy human volunteers (in this case the investigators themselves) resulted in the stimulation of thyroid hormone production Citation[4]. Subsequent studies showed that the target of LATS was the TSHR. TSHR is normally activated by thyroid stimulating hormone (TSH, also called thyrotropin), a product of the pituitary gland, which regulates the production and secretion of thyroid hormones from the thyroid gland. TSHRs are not exclusively expressed in thyroid tissue, as originally thought, but are also present on a number of extrathyroid tissues including lymphocytes, thymus, pituitary, testes, kidney, brain, adipose tissue, fibroblasts and bone. Recent evidence indicates that TSH regulates lipolysis, osteoclast activity and other areas of mammalian physiology Citation[2].
Evidence for autoimmunity
The seminal studies of Adams and colleagues showed that the serum immunoglobulin fraction from individuals with Graves’ disease could stimulate radioiodine release from prelabeled guinea pig thyroid tissue. In a classic adoptive transfer experiment they showed that infusion of the thyroid stimulating fraction into healthy human volunteers (themselves) resulted in increased thyroid hormone production Citation[4,5]. Thus, the pioneering experiments of Adams and colleagues provided the first clear evidence that Graves’ disease is an autoimmune disease. Another form of adoptive transfer occurs naturally during pregnancy when the mother has Graves’ disease. In this case the transfer of maternal TSHR antibodies to the fetus can cause the fetus to have a transient hyperthyroidism, which goes away following pregnancy as maternal antibody in the neonatal circulation decreases Citation[6].
Evidence for TSHR as an autoantigen
It took nearly 20 years before the TSHR was identified as the target of the LATS activity initially discovered by Adams. Evidence for binding to TSHR was provided by Smith and Hall who showed that IgG from patients with Graves’ disease competed for bovine TSH binding to porcine thyroid cell membranes Citation[7]. The ability of patient IgG to compete for TSH binding to the TSHR formed the basis of clinical assays using recombinant human TSHR-coated tubes Citation[8]. Improvements in the competition assays came with the availability of monoclonal antibodies to the TSHRs. With these highly specific monoclonal antibody reagents it was possible to detect TSHR antibodies based on competition with the monoclonal antibodies. The most recent advance in the monoclonal antibody category came with the isolation of a human monoclonal antibody from peripheral blood lymphocytes of a 19-year-old male with Graves’ disease Citation[9]. This monoclonal antibody binds to the TSHR with high affinity and is a potent inhibitor of TSH binding to the TSHR. Additionally the monoclonal antibody, termed ‘M22’, is a potent stimulator of cyclic AMP production in Chinese hamster ovary cells expressing the TSHR. The use of M22 as a labeled ligand in competition with individual patient TSHR autoantibodies has enabled estimation of their serum concentrations and affinities. TSHR autoantibodies in Graves’ disease patients range from 5 to 60 ng/ml or 5 to 60 ppm of total serum IgG Citation[10,11]. Treatment of Graves’ disease with rituximab, a B-cell depleting monoclonal antibody directed to a cell surface antigen present on B cells (CD20), specifically reduces the production of TSHR antibodies Citation[12].
In addition to binding assays based on competition with TSH or monoclonal antibodies, it has also been possible to develop bioassays that directly measure the ability of IgG fractions from patients with Graves’ disease to activate the TSHR Citation[8]. Bioassays originally relied on the detection of cAMP production in cell lines that normally express the TSHR or had been genetically modified to do so. More recent modifications have involved the introduction of luciferase reporter genes equipped with cAMP response elements. For these bioassays, only a small amount of patient sera is required to detect TSHR autoantibodies based on increased luciferase expression. Unlike the binding competition assays described previously, the bioassays have the ability to discriminate between agonistic and antagonist autoantibodies.
Structure of TSHR & TSHR autoantibody complex
The TSHR belongs to a subfamily of GPCRs (including receptors for luteinizing hormone and follicle stimulating hormone) that are characterized by a large, extracellular, leucine-rich repeat domain (LRD) that constitutes the A-subunit of the receptor Citation[1]. The LRD (amino acids 1–315) is derived by proteolytic processing from the initial full-length receptor (764 amino acids) following removal of a 50-amino acid extracellular segment from 316 to 366. As a result of this proteolytic processing, the mature receptor consists of two subunits bound together by disulfide bonds. The A-subunit consists of nine leucine-rich repeat domains and N- and C-terminal cysteine-rich regions. The B-subunit consists of seven transmembrane helices and a short cytoplasmic tail. In an effort to understand how Graves’ disease autoantibodies activate the TSHR, Sanders and colleagues isolated a human monoclonal antibody from lymphocytes of a patient with Graves’ disease and studied the interaction of this autoantibody with the receptor Citation[13–15]. They found that the autoantibody and its Fab fragment bind to the TSHR with high affinity, inhibit labeled thyrotropin binding to the receptor and stimulate cAMP production in Chinese hamster ovary cells genetically engineered with TSHRs. For a more detailed analysis of antibody–receptor interaction, they prepared a complex consisting of the Fab fragment of the autoantibody (M22) and the extracellular domain of the TSHR (specifically the first 260 amino acids of the LRD) Citation[15]. Crystals suitable for x-ray diffraction analysis were obtained and the structure solved at 2.55 Å resolution. The results show that the antibody binds extensively to the concave surface of the LRD in a way that is believed to be nearly identical to that of the natural ligand. Thus, the structural analysis of autoantibody binding to the external domain of TSHR suggests that antibody-mediated receptor activation is accomplished in a way similar to that of the natural ligand, TSH. However, the mechanism by which TSH or autoantibody binding to the LRD initiates transmission of a signal to the B-subunit (the transmembrane domain) followed by heterotrimeric G-protein activation is not known.
Animal models
Initial attempts to generate a Graves’ disease animal model used recombinant human TSHR ectodomain protein produced in bacterial or eukaryotic expression systems. The resulting TSHR antibodies blocked TSH-mediated TSHR activation, resulting in mice with hypothyroidism. Successful efforts to produce a mouse model of Graves’ disease (i.e., hyperthyroidism) involved injecting mice with cDNA Citation[16] or adenoviruses encoding TSHR Citation[17]. The resulting mice were characterized by thyroid hypertrophy and the presence of autoantibodies that activated the TSHR. The success of these experiments is presumably based on the fact that TSHR peptides need to be presented to T cells via MHC class II molecules to elicit production of autoreactive T cells. These studies suggested that it was necessary to allow cells to produce intact receptors to maintain conformational integrity. These studies also showed that if mice could be immunized in such a way as to produce TSHR-activating autoantibodies the mice would have features of Graves’ disease.
β1-adrenergic receptor agonistic autoantibodies & the pathogenesis of dilated cardiomyopathy
Pathogenic potential of β1-adrenergic receptor agonistic autoantibodies
Dilated cardiomyopathy (DCM) is a heart muscle condition characterized by a progressive loss of cardiac function associated with dilation of the left ventricle. In approximately 60% of cases the etiology is unknown and the condition is referred to as idiopathic DCM. The condition represents the main cause for severe heart failure and disability in young adults. It is also prevalent among individuals with Chagas’ disease, an endemic parasitic disease of Latin America (see following paragraphs).
Very early evidence by Sterin-Borda and colleagues showed that sera of patients with Chagas’ disease cardiomyopathy contain IgG that binds β1-adrenergic receptors of the heart and stimulates contractile activity of the myocardium Citation[18]. This activity is inhibited by β1-adrenergic receptor antagonists but not by β2-adrenergic receptor antagonists. Subsequent studies showed that these β1-adrenergic receptor autoantibodies were not restricted to Chagas’ cardiomyopathy but were frequently present in individuals with idiopathic DCM. The β1-adrenergic receptor is a member of the superfamily of GPCRs, whose main function in the heart is to positively regulate beating rate and beating strength Citation[19]. These receptors are also the predominant adrenoceptor subtype in the heart. β1-adrenergic receptor (AR)-agonistic autoantibodies (AAs) activate the β1-AR signaling cascade and are associated with poor left ventricular function, a higher prevalence of serious ventricular arrhythmias and a higher incidence of sudden death.
Numerous biological and immunological studies indicated that β1-AR-AAs are directed to epitopes on the first or second extracellular loop of the β1-adrenergic receptor Citation[20–27] and together accounted for 70–80% of idiopathic DCM. According to evidence from two groups Citation[26,27] approximately half of the autoantibodies were directed to each extracellular loop. More recently, the use of a newly developed biological assay based on the use of a sensitive fluorescence resonance energy transfer assay to detect intracellular cAMP showed that antibodies directed to the first extracellular loop are not as active as those directed to the second extracellular loop Citation[26]. Nevertheless, the enhanced sensitivity of this assay allowed both types of antibodies to be reliably detected. According to this assay, functionally active (i.e., stimulatory) β1-AR-AAs are present in less than 1% of a population of healthy individuals Citation[19]. By contrast, agonistic autoantibodies to the β1-AR were observed in approximately 20% of patients with ischemic cardiomyopathy and approximately 70% of patients with DCM. Overall, available data suggest that the cardiomyopathic phenotype results from a mild but sustained receptor activation. Additional evidence for the pathogenic nature of these autoantibodies comes from animal model studies described later.
Chagas’ disease
Chagas’ disease is an endemic parasitic disease of Latin American countries and is caused by infection with the flagellate protozoan, Trypanosoma cruziCitation[28]. Following a long latency period of 10–30 years, nearly 30% of infected individuals develop life-threatening cardiomyopathies associated with arrhythmias, heart failure and frequently sudden death. Chagas’ heart disease is a major cause of morbidity and mortality in Latin America. Recent evidence suggests an autoimmune contribution to Chagas’ pathophysiology, especially Chagas’ heart disease Citation[29]. This view is supported by evidence that nearly all Chagas’ patients with cardiomyopathy have agonistic autoantibodies to cardiac GPCRs, including β1-AR, β2-AR and the M2 muscarinic receptor Citation[30]. These autoantibodies are absent from control, uninfected individuals and present in approximately 30% of asymptomatic T. cruzi-infected individuals. Molecular mimicry between an immunodominant highly charged pentapeptide epitope on ribosomal protein P0 of T. cruzi and a highly conserved site on the second extracellular loop of the β1-AR has been proposed as the basis for autoantibody production Citation[31]. In this regard, it is interesting to note that the β1-AR-activating autoantibodies associated with Chargas’ disease only recognize the epitope on the second extracellular loop Citation[32]. Activating antibodies directed to the first extracellular loop are not observed in Chagas’ disease cardiomyopathy, a finding that is consistent with the molecular mimicry hypothesis.
Numerous studies suggest that, as with idiopathic DCM, β1-AR-AAs contribute to Chagas’ heart disease. Both patient groups carry a high percentage of β1-AR-AA and the transfer of β1-AR-AA to experimental animals resulted in a dilated cardiomyopathy similar to that observed in humans Citation[33,34]. At the cellular and subcellular levels, changes in the action potential duration and contractility of cardiomyocytes have been observed following the addition of autoantibodies Citation[35]. Because of these experiments, Chagas’ heart disease is increasingly considered an autoimmune disease, where agonistic autoantibodies to GPCRs such as the β1-ARs, β2-ARs and M2-muscarinic receptors contribute to disease pathogenesis Citation[28]. It has been shown for patients with DCM, and suggested for patients with Chagas’ cardiomyopathy, that removal of these pathogenic autoantibodies would yield significant clinical benefit. These autoantibodies may serve as presymptomatic markers to identify patients at risk for subsequent development of Chargas’ cardiomyopathy.
Animal models
Realizing that agonistic autoantibodies to the β1-ARs were commonly targeted to the second extracellular loop (ECII) of the receptor, Jahns and colleagues took an experimental approach in rats to demonstrate the pathogenic potential of such autoantibodies Citation[33]. They immunized rats with a fusion protein encoding the ECII of the β1-AR. They observed that the immunized rats developed agonistic autoantibodies to the β1-AR and that the appearance of these autoantibodies was accompanied with cardiac dilatation and dysfunction, finally resulting in a full DCM phenotype. Direct evidence for a pathogenic role of β1-AR agonistic autoantibodies was provided by adoptive transfer experiments in which DCM was produced in healthy rats following isogenic transfer of autoantibodies from rats immunized with β1-AR ECII sequences. The introduction of anti-β1-AR-ECII agonistic antibodies resulted in a cardiomyopathy phenotype characterized by progressive left ventricular dilatation and dysfunction, a relative decrease in left ventricular wall thickness and selective downregulation of β1-ARs, all features seen in human DCM. These results suggest that the induced and transferred DCM phenotype can be attributed to the mild but sustained receptor activation resulting from stimulatory anti-β1-AR-ECII antibodies. A large clinical diagnostic study is underway to evaluate the role of β1-AR agonistic autoantibodies in heart disease Citation[36] using a newly developed functional assay discussed previously Citation[26].
α1-adrenergic receptor-activating autoantibodies & refractory hypertension
Background
α1-adrenergic receptors (α1-ARs) regulate several important cardiovascular actions. They are predominantly located postsynaptically on vascular smooth muscle cells (VSMCs), where they are targets of circulating norepinephrine and regulate VSMC contraction . Their presence on cardiomyocytes controls cardiac inotropy, hypertrophy and remodeling. Initial evidence that the α1-AR was the target of agonistic autoantibodies was reported many years ago by Fu et al., who showed that α1-AR-AA were present in seven of 11 patients with secondary hypertension, three of 15 patients with malignant hypertension and four of 33 healthy controls Citation[37]. A follow-up study examined immunoglobulin fractions in 54 severely hypertensive patients and found that α1-AR-AAs were present in 44% of these patients Citation[38]. However, such autoantibodies were also seen in 12% of normotensive control subjects. More recently, Wenzel et al. examined a larger cohort of patients with refractory hypertension (defined as requiring three or more hypertensive medications) and observed α1-AR-AAs in 41 of 81 patients Citation[39]. All of these studies showed that α1-AR-AA stimulated contraction rates by cultured neonatal cardiomyocytes, a property that is blocked by the presence of prazosin, a α1-AR antagonist. Additionally, Yan et al. showed that these autoantibodies provoked increased contraction of thoracic aortic rings in vitro and increased blood pressure when injected into rats Citation[40].
Pathologic potential of α1-AR-AAs
A variety of experimental approaches were used to evaluate the pathologic potential of α1-AR-AAs Citation[39]. For this purpose, these antibodies were purified from hypertensive patients by affinity chromatography using a peptide corresponding to the ECII of the α1-AR. Surface plasmon resonance measurements showed that the purified antibody displayed a high binding affinity (Kd˜50 nM) and was specific for sequences of the ECII. Functional analysis of the purified autoantibody showed that it gave a dose-dependent stimulation of the beating rate of neonatal rat cardiomyocytes, which was inhibited by the α1-AR antagonist prazosin Citation[39]. Additional function studies showed that these autoantibodies stimulated contraction of mesenteric arteries and stimulated intracellular Ca2+ mobilization in neonatal cardiomyocytes. Analysis of key intracellular signaling molecules revealed autoantibody-mediated activation of protein kinase C-α and transient phosphorylation of ERK1/2. Overall, these data provide strong support for the concept that these α1-AR-AAs have the potential to contribute to hypertension in the patients from whom they were obtained Citation[39].
The potential contribution of α1-AR-AAs to hypertension was tested by the use of immunoadsorption to remove these antibodies from five patients with refractory hypertension Citation[39]. Treatments were performed daily for 5 days and the cardiomyocyte contraction assay was used to evaluate removal of α1-AR-AAs. The results showed that the pathological autoantibodies were successfully removed from treated patients, reaching the normal range by 5 days of immunoadsorption. The reduced α1-AR-AA activity persisted for the 180-day period of the trial. Blood pressure measurements showed a significant reduction in mean arterial pressure by 5 days and remained low for the 180-day observation period. These results suggest that α1-AR-AAs are of potential pathophysiological relevance and may contribute to the development of refractory hypertension. Thus, the results from this small and uncontrolled clinical study suggest that the removal of α1-AR-AAs by immunoadsorption lowers blood pressure in α1-AR-AA-positive patients with intractable hypertension. Overall, these results suggest that α1-AR AAs represent promising therapeutic targets in the medical management of refractory hypertension.
Animal models
Very recent studies from the Dechend group have used animal models to show that α1-AR-AAs have the ability to contribute to cardiac end organ damage Citation[41]. For these studies, Lewis rats were immunized with the second loop peptide and monitored for changes in blood pressure and cardiovascular function. The immunized rats developed α1-AR-AAs that were detected by the cardiomyocyte contraction assay and by ELISA. These antibodies were capable of initiating relevant intracellular signaling pathways as judged by ERK1/2 phosphorylation. The appearance of α1-AR-AAs was followed by diastolic dysfunction and hypertrophy in immunized rats. Genes encoding sarcomeric proteins, collagen, extracellular matrix proteins, calcium regulating proteins and proteins of energy metabolism in immunized rat hearts were upregulated, compared to controls. Furthermore, fibrosis was present in the hearts of immunized animals and not controls. Additional studies by Karczewski et al. have raised α1-AR-AAs by immunizing rats with second loop peptides and have shown that these antibodies stimulate the mobilization of intracellular calcium stores Citation[42]. These preclinical studies suggest that α1-AR-AAs may contribute to cardiovascular end-organ damage and hypertension.
Angiotensin receptor-activating autoantibodies & preeclampsia
Background
Preeclampsia is a life-threatening hypertensive complication of pregnancy that is a leading cause of maternal and neonatal mortality and morbidity in the USA and the world Citation[43]. It is a multisystem disorder generally appearing after the 20th week of gestation and characterized by hypertension, proteinuria, inflammation, coagulopathy and is often associated with intrauterine growth restriction. The condition affects approximately 7% of first pregnancies and accounts for over 80,000 premature births each year in the USA (˜15% of total premature births), over US$4 billion in medical costs Citation[44] and immeasurable human suffering. Available strategies to manage preeclampsia are inadequate and reflect a lack of fundamental understanding of the etiology and pathogenesis of the disorder.
Pathological potential of angiotensin receptor agonistic autoantibodies
Numerous studies over the past 12 years have shown that women with preeclampsia possess autoantibodies, which bind to and activate the major angiotensin II receptor (AT1) on many cell types Citation[45–49]. These autoantibodies, termed ‘AT1-AAs’, activate AT1 receptors on cardiac myocytes resulting in increased contraction rates Citation[50]. AT1-AAs stimulate the synthesis of NADPH oxidase in several cell types, including VSMCs and trophoblasts cells, resulting in increased production of reactive oxygen species (ROS) and oxidative damage Citation[51]. Autoantibody-induced synthesis of tissue factor by vascular smooth muscle cells and monocytes may contribute to hypercoagulation often associated with preeclampsia Citation[52]. The activation of AT1 receptors on mesangial cells and trophoblasts results in increased synthesis and secretion of plasminogen activator inhibitor-1, which is elevated in women with preeclampsia and may also contribute to hypercoagulation Citation[53,54]. These autoantibodies also stimulate production of soluble fms-like tyrosine kinase-1 (sFlt-1; a soluble form of the VEGF receptor-1) and soluble endoglin (sEng; a soluble form of the TGF-β receptor) by the placenta Citation[55,56]. These anti-angiogenic factors are elevated in women with preeclampsia and believed to contribute to vascular and renal symptoms of the disease. AT1-AAs also stimulate vasoconstriction of rat thoracic aorta Citation[57]. Thus, a large body of evidence indicates that AT1-AAs activate AT1 receptors on a variety of cells and provoke biological responses that may contribute to the pathophysiology of preeclampsia .
Because these earlier studies were restricted to the use of in vitro cultured cell systems or tissue explants, they did not directly address the relevance of AT1-AAs to hypertension and proteinuria, the defining clinical features of preeclampsia. For this reason, an adoptive transfer in vivo experimental approach was used to show that the introduction of these autoantibodies into pregnant mice resulted in hypertension, proteinuria and a variety of other features of preeclampsia including increased production of sFlt-1 and sEng Citation[58]. Proteinuria is one of the defining clinical features of preeclampsia and is accompanied by a characteristic renal abnormality termed glomerular endotheliosis (glomerular endothelial cell swelling). Both of these features appeared in pregnant mice following AT1-AA injection and were prevented by coinjection with losartan or a 7-amino acid epitope peptide that corresponds to a site on the ECII of the AT1R. These results indicate that proteinuria and renal pathology result from autoantibody-induced AT1 receptor activation in pregnant mice. Because these autoantibodies produce clinical features of preeclampsia in pregnant mice, it is likely that they contribute to symptoms of preeclampsia in the women from whom they were obtained. This view is supported by data showing that AT1-AAs are highly prevalent in preeclampsia and that antibody titers correlate to the severity of the disease Citation[59]. Thus, in vitro and in vivo findings with patients’ sera suggest a pathophysiological role for these autoantibodies in preeclampsia and provide experimental support for the hypothesis that preeclampsia is an autoimmune condition characterized by the presence of disease-causing autoantibodies.
The contribution of AT1-AA to intrauterine growth restriction associated with preeclampsia
Preeclampsia is often associated with intrauterine growth restriction (IUGR) Citation[60]. Growth-restricted fetuses have a higher incidence of mortality and morbidity than fetuses of normal growth and are at increased risk for future development of metabolic disorders such as hypertension, coronary heart disease, dyslipidemia, obesity, impaired glucose tolerance, Type 2 diabetes mellitus and many other diseases Citation[61–63]. Recently, Irani et al. reported that AT1-AA are present in the cord blood of women with preeclampsia and retain the ability to activate AT1 receptors Citation[64]. Using an adoptive transfer model of preeclampsia in pregnant mice, they showed that AT1-AAs cross the placenta and enter the fetal circulation where they are associated with small fetuses with renal and hepatic abnormalities. AT1-AAs also induce apoptosis in the placentas of pregnant mice, human villous explants and human trophoblast cells in culture Citation[64]. Finally, autoantibody-induced IUGR and placental apoptosis are diminished by either losartan or the autoantibody-neutralizing 7-amino acid epitope peptide. These studies highlight AT1-AA as a potential contributor to preeclampsia-associated IUGR and offer two possible underlying mechanisms: a direct detrimental effect on fetal development by crossing the placenta and entering the fetal circulation, and fetal growth restriction secondary to AT1-AA-induced placental damage.
Animal models
As discussed previously, preeclampsia in humans is characterized by the presence of autoreactive antibodies, AT1-AA, that are likely to contribute to the pathophysiology of the disease Citation[58]. These antibodies have also appeared in experimentally induced animal models of preeclampsia initiated by placental ischemia or the infusion of inflammatory cytokines. Granger and colleagues developed a rat model of preeclampsia based on surgically induced placental ischemia resulting from reduced uterine perfusion pressure (RUPP) Citation[65,66]. These surgically manipulated pregnant rats develop hypertension, proteinuria and other features of preeclampsia, including elevated levels of sFlt-1, sEng, increased inflammatory cytokines such as TNF-α and IL-6, increased endothelin-1 (ET-1) production and endothelial dysfunction Citation[67,68]. Remarkably, RUPP rats also develop AT1-AA Citation[69–71]. RUPP-induced hypertension is inhibited by losartan but not by an angiotensin-converting enzyme (ACE) inhibitor, suggesting that AT1-AAs contribute to hypertension observed in this animal model of preeclampsia. LaMarca and colleagues Citation[69] found that low dose TNF-α infusion into pregnant rats also resulted in increased blood pressure and the appearance of AT1-AA. Neither RUPP manipulation or TNF-α infusion caused hypertension in nonpregnant rats, suggesting a role for pregnancy and the placenta in hypertension and the production of AT1-AAs. Treatment of RUPP animals with rituximab, to prevent the mobilization of B lymphocytes inhibited the production of AT1-AA and reduced hypertension Citation[71]. A role for CD4+ T helper cells in RUPP-induced hypertension and AT1-AA production has also been demonstrated Citation[72].
AT1-AA also appear in a transgenic model of preeclampsia in pregnant rats based on overproduction of Ang II during pregnancy Citation[73]. AT1R-activating antibodies have been raised in rabbits based on immunization with the dominant 7-amino acid epitope present on the ECII of the receptor. Active immunization with AT1R second loop peptides has also been used to obtain high titers of AT1R antibodies in rats and was associated with endothelial cell disfunction Citation[74]. AT1R activating antibodies from rats and rabbits, as with those from women with preeclampsia, behave as functional mimics of Ang II and cause widespread activation of AT1Rs when introduced into laboratory animals. Adoptive transfer studies in pregnant mice and/or rats have demonstrated that autoantibody-mediated receptor activation results in increased production of proinflammatory cytokines (TNF-α, IL-6) Citation[75,76], antiangiogenic factors (sFlt-1, sEng) Citation[55,77] and a potent vasoconstrictor (ET-1) Citation[70,76]. Pharmacologic experiments with receptor antagonists or neutralizing antibodies have shown that the antibody-mediated induction of each of these compounds contributes to clinical features of preeclampsia in animal models. Additional adoptive transfer studies in pregnant rats indicate that AT1-AAs contribute to placental oxidative stress Citation[78], renal endothelial dysfunction Citation[79] and increased Ang II sesnsitivity Citation[48]. Antibody-induced pathophysiology is blocked by the 7-amino acid epitope peptide or an AT1 receptor antagonist, indicating that autoantibody-induced pathophysiology is mediated by AT1R activation.
Thus, a large number of publications from multiple groups report the results of adoptive transfer and immunization experiments showing the pathological potential of AT1-AA. Excessive AT1 receptor activation, resulting from the combined action of Ang II and AT1-AA, represents a unifying explanation for many of the pathophysiological features of preeclampsia. Preeclampsia is not currently considered an autoimmune condition. However, a growing body of research implicates AT1-AAs in the pathogenesis of preeclampsia and is likely to provide new opportunities for screening, diagnosis and treatment Citation[47,80]. Currently, the factors that contribute to the production of AT1-AAs and the autoimmune recognition of the AT1 receptor peptide epitope are not well understood and could be associated with hypoxia-mediated vascular injury in the placenta. Furthermore, the generation of these autoantibodies may be secondary to the placental ischemia, vascular damage and increased maternal inflammatory response associated with preeclampsia. Promising experimental approaches to study the relationship between preeclampsia and immunity include the experimental induction of uteroplacental ischemia and the infusion of inflammatory cytokines Citation[80].
The role of AT1-AA in renal allograft vascular rejection
Initial clinical evidence
Despite accurate tissue typing and novel immunosuppressive modalities, graft rejection remains a constant threat to allograft survival in kidney transplant recipients. A particularly aggressive form of graft rejection is due to antibody-mediated rejection, a process featuring acute vascular lesions in the allograft including thrombosis, fibrinoid necrosis and endarteritis. Donor-specific anti-HLA alloantibodies initiate complement mediated and antibody-dependent cell-mediated cytotoxicity in approximately half of these cases. The accumulation of the complement degradation product, C4d, is regarded as a marker for an antibody-mediated alloresponse and is associated with poor graft survival. However, nearly half of antibody-mediated allograft rejections do not involve anti-HLA antibodies, but instead are targeted to other antigens and include self antigens and donor alloantigens. The identification of non-HLA antigens that are relevant to rejection may provide insight into underlying mechanisms, define risk-related features and facilitate the development of specific therapies. Dragun et al. investigated a group of 33 kidney-transplant recipients who had refractory vascular rejection Citation[81–83]. A total of 13 of these patients had donor specific anti-HLA, whereas 20 did not. Of the latter, 16 were characterized by malignant hypertension, four of whom had seizures. Acute vascular rejection and malignant hypertension occurred during the first week after transplantation. The combination of vascular pathology, hypertension and seizures prompted these investigators to consider the presence of AT1-AA, a pathogenic autoantibody they had previously observed in women with preeclampsia, a condition also associated with vascular lesions, hypertension and seizures. The cardiomyocyte contraction assay was used to show that AT1-AAs were present in all 16 patients with malignant hypertension and in none of the remaining patients. Retrospective analysis of historic sera taken from patients prior to transplantation revealed the presence of AT1-AA in all 16 patients presenting with acute vascular rejection and malignant hypertension during the week following transplantation. Subsequent analysis of 278 kidney transplant recipients performed at their center revealed a crude prevalence of AT1-AA associated vascular rejection episodes of 3.6% (i.e., ten patients). Consistent with these findings are the reports of others indicating the presence of AT1-AA in some patients with essential hypertension Citation[84–87].
Antibody characterization
The receptor-activating autoantibodies belonged to the IgG1 and IgG3 subclasses and interacted with two different epitopes on the ECII of the AT1R, one of which was identical to the epitope identified in preeclampsia. Tissue factor expression was increased in renal-biopsy specimens from patients with AT1-AA, reminiscent of that seen in placental biopsies of patients with preeclampsia. Autoantibody-binding to AT1R in vascular cells activated downstream signaling pathways including phosphorylation of ERK1/2 kinase, NF-κB and AP-1. Passive antibody transfer into a rat kidney transplantation model induced vasculopathy and hypertension. In vitro and in vivo effects of AT1-AA were prevented by the AT1R antagonist, losartan. These results suggest that antibody-mediated AT1R activation contributes to refractory vascular rejection and that affected patients may benefit from removal of the pathogenic autoantibodies or blockade of AT1Rs, or both.
Therapeutic approaches to reduce impact of pathogenic autoantibodies
Seven of the 16 patients originally identified with AT1-AAs were treated with a combination therapy consisting of plasmaphoresis, intravenous immunoglobulin infusions and the AT1R blocker, losartan. This treatment regimen resulted in improved renal function and graft survival compared to AT1-AA-positive patients who received standard treatment for humoral rejection. The finding that patients with preformed AT1-AA develop severe vascular rejection that can be treated with specific therapy suggests that pretransplant testing for this autoantibody may help to improve individual risk assessment and therapeutic options offered to potential transplant recipients Citation[88,89].
Functional autoantibodies to vascular receptors in systemic sclerosis
Background
Systemic sclerosis (SSc) is a chronic systemic autoimmune disease (primarily of the skin) characterized by fibrosis, vascular alterations, and autoantibodies. There are two major forms. Limited SSc is characterized by cutaneous involvement mainly affecting the hands, arms and face. Pulmonary arterial hypertension occurs in some patients and is the most serious complication for this form of the disease. Diffuse SSc is a rapidly progressing form of the disease and affects large areas of the skin and one or more internal organs (kidneys, esophagus, heart or lungs). Difusse SSc can be quite disabling. The prognosis is generally good for patients with limited SSc who escape pulmonary complications. The situation is worse for individuals with the diffuse disease, particularly those in older age groups, and for males. Death occurs most often from pulmonary, cardiac or renal complications, secondary to hypertension. ACE inhibitors, ET-1 receptor blockers and AT1R blockers reduce hypertension and alleviate some of the cardiac, renal and pulmonary manifestations of SSc. The combination of autoimmunity, vasculopathy and hypertension resembled conditions present in preeclampsia and prompted Dragun and colleagues to consider the possibility that receptor-activating autoantibodies were involved Citation[90].
Evidence for receptor-activating autoantibodies
To test their hypothesis, Dragun and colleagues developed a cell-based ELISA to detect antibodies to the Ang II receptor, AT1 and the ET-1 receptor, ETA. Solid phase sandwich ELISAs were constructed using extracts from Chinese hamster ovary cells that overexpress human AT1R or ETAR and antibody binding was conducted under conditions designed to preserve sensitive conformational epitopes. Their results indicated that most SSc patients possess antibodies to each of these receptors Citation[90]. Specific binding of autoantibodies to AT1Rs and ETARs on human microvascular endothelial cells (HMECs) was demonstrated by coimmunoprecipitation. Antibodies to each receptor are biologically active in that they induce receptor-directed ERK1/2 phosphorylation and increased TGF-β gene expression in HMECs. Ang II and ET-1 induce collagen synthesis via target receptor stimulation in fibroblasts, features that could be attributed to the vascular receptor-activating autoantibodies. The biological effects of both autoantibodies were blocked by the respective receptor antagonists, providing additional evidence for antibody-mediated receptor activation. Higher antibody titers were associated with diffuse SSc and for the presence of late complications such as pulmonary hypertension, lung fibrosis and digital ulcers. Such patients also had a higher chance of dying from SSc complications. Thus, according to these authors, AT1R and ETAR agonistic autoantibodies may contribute to disease pathogenesis in SSc and link disease features including autoimmunity, vascular pathology and hypertension.
ETAR-activating autoantibodies & pulmonary arterial hypertension
The presence of ETAR-activating autoantibodies has also been reported for patients with pulmonary arterial hypertension (PAH) Citation[91–93]. According to results presented in these preliminary reports, the vast majority of patients with idiopathic PAH tested positive for agonistic autoantibodies to the ETAR and/or the α1-adrenergic receptors. Initial efforts to remove these pathogenic autoantibodies by immunoadsorption showed encouraging short-term results.
Discussion
The burden of autoimmune diseases
The immune system is adapted to identify and eliminate foreign antigens and the organisms bearing them. This process is usually carried out in two overlapping phases representing initial activation of the innate branch of the immune system followed by activation of the adaptive immune system. Activation of the innate system is usually accompanied by an inflammatory response that persists until the foreign antigen is eliminated and the damaged tissue repaired. However, in the case of autoimmunity, the target autoantigen is rarely eliminated and the adaptive arm of the immune response continues unabated resulting in tissue injury and inflammation. Autoimmune diseases are sometimes categorized as organ specific (e.g., Type I diabetes, Graves’ disease, multiple sclerosis) or systemic (e.g., rheumatoid arthritis, systemic lupus erythematosus, SSc). Autoimmune diseases can also be categorized as humoral or cellular depending on whether disease pathology is largely dependent on autoantibodies (e.g., Graves’ disease, myasthenia gravis, systemic lupus erythematosus) or cytotoxic T cells (Type I diabetes, multiple sclerosis, rheumatoid arthritis). It is important to note that most of the diseases just referred to were not originally recognized as autoimmune diseases and only came to be understood as such following years (often decades) of investigation. Of the diseases considered in this article, Graves’ disease and SSc are universally considered to be autoimmune diseases. The others, including idiopathic dilated cardiomyopathy, Chagas’ disease, malignant hypertension (with and without renal allograft rejection) and preeclampsia are still under investigation as autoimmune diseases Citation[94]. Although SSc has long been considered an autoimmune disease, the contribution of receptor-activating autoantibodies to disease pathology is a matter of recent consideration Citation[90].
Therapeutic approaches to renal allograft rejection & refractory hypertension
The research reviewed here provides examples of autoantibody-mediated receptor activation that likely contribute to disease. Examples of therapeutic removal and receptor blockade were provided for kidney transplant recipients suffering from renal allograft rejection and malignant hypertension characterized by the presence of AT1R activating autoantibodies Citation[82,83,88]. Five patients selected for this treatment protocol had significantly improved graft survival compared to autoantibody-positive patients receiving standard therapy. A related treatment protocol was carried out for five patients with refractory hypertension associated with α1-AR-activating autoantibodies Citation[39]. In this case, immunoadsorption was used to remove the pathogenic autoantibodies on each of 5 consecutive days, a process resulting in normalization of blood pressure. In each case, these studies showed that it was possible to effectively remove pathogenic autoantibodies with resulting therapeutic benefit. These preliminary, noncontrolled clinical studies provide promising evidence that therapeutic removal of pathogenic receptor-activating autoantibodies may have significant clinical benefit. Pharmacologic intervention with specific receptor antagonists is also appropriate in these cases. However, teratogenic concerns may limit the use of receptor blockers during pregnancy. Receptor blockers, in general, may have unwanted side effects, whereas antibody removal or neutralization should not. Knowledge of the specific epitope, offers possible approaches for blocking the autoantibody and preventing antibody-mediated receptor activation.
For maximum therapeutic benefit, it will be preferable to identify the pathogenic autoantibodies before the onset of clinical symptoms. This approach is already underway for renal transplant recipients where 3–4% of candidates for renal transplant are positive for AT1R autoantibody Citation[89], findings consistent with the presence of these autoantibodies in individuals with essential hypertension Citation[84–87]. Evidence indicates that these patients will benefit from more specialized treatment protocols before and immediately following transplantation.
Presymptomatic screening for preeclampsia
Research reviewed here suggests that preeclampsia may be a pregnancy-induced autoimmune condition characterized by the presence of pathogenic autoantibodies, AT1-AAs. At this time, we only have data pertaining to the presence and abundance of AT1-AAs at the time of diagnosis for women with preeclampsia and at the time of delivery for normotensive pregnant women. Previously published data of Walther et al.Citation[95] indicate that women with abnormal Doppler ultrasound, an early indicator of reduced placental perfusion, possess AT1-AAs well ahead of the appearance of clinical symptoms of preeclampsia. These data are limited to a small number of patients, but suggest that production of AT1-AA tracks with impaired placental perfusion and represents a risk factor for subsequent development of preeclampsia. An important component to the management of preeclampsia would be a sensitive and reliable immunological test to identify the onset of AT1-AA production during pregnancy to determine the temporal relationship between the onset AT1-AA production and the appearance of clinical symptoms of preeclampsia. Such studies are likely to identify AT1-AA as a significant presymptomatic risk factor for preeclampsia and provide a window of therapeutic opportunity to block or remove these pathogenic autoantibodies before they initiate clinical symptoms.
Most research reports dealing with AT1-AA have relied on biological assays based on increased cardiomyocyte contraction Citation[50] or increased expression of a reporter gene Citation[59] to detect AT1-AA. The use of ELISAs for the detection of AT1-AA in patients with preeclampsia has been reported Citation[57] but has not yet been used in a prospective study to determine usefulness as a presymptomatic test. A cell-based ELISA has been developed for detection AT1-AA in patients with SSc Citation[90] and renal allograft vascular rejection Citation[89] and may be useful for detecting AT1-AA in patients with preeclampsia. A successful effort to develop sensitive and reliable high-throughput screening of maternal serum for the presence of AT1-AA would likely have significant applications in obstetrical medicine.
Factors contributing to autoantibody production
It is not a surprise that the autoantibodies discussed in this article are directed to extracellular domains of their target receptors. For Graves’ disease, the autoantibodies are directed to the unusually large N-terminal A fragment of the thyrotropin receptor Citation[15]. This fragment is derived from the full-length receptor by proteolytic cleavage and remains associated with the receptor following cleavage. Some investigators have speculated that the A fragment subunit is a better autoantigen than the full-length receptor Citation[96,97]. Other autoantibodies considered in this article are directed at the second (or central) extracellular loop of their target GPCR Citation[94]. The immunological basis for the predominance of the ECII is not understood but may be related to molecular mimicry as discussed later.
For autoantibodies to be produced against extracellular domains of GPCRs it is necessary to have a loss of self tolerance for the humoral (antibody-mediated) and cellular (T-cell mediated) arms of the immune system. In order to activate T cells, GPCRs must be degraded to small oligopeptides by antigen-presenting cells, such as macrophages or dendritic cells, and presented to T cells by HLA class II molecules. Such a process could occur as a result of macrophage engulfment of damaged cells. For autoantibody production to occur, it is necessary that an extracellular domain of a GPCR be recognized by B-cell receptors on the surface of a B lymphocyte. With the assistance of a cognate helper T-cell, the autoantigen stimulated B cells could proliferate and produce autoantibody. Factors accounting for the breakdown of self tolerance in this scenario are not understood, but may involve molecular mimicry with a previously encountered pathogen. Evidence for molecular mimicry has been provided for the generation of β1-AR-AAs, where involvement of T. cruzi infection is associated with Chagas’ cardiomyopathy Citation[31]. In this case, a pentapeptide sequence on the ECII of the β1-AR matches an immunological epitope on a T. cruzi ribosomal protein. Molecular mimicry may also be involved in the generation of AT1-AAs associated with preeclampsia Citation[49,98], although the issue is unresolved Citation[99]. In this case, the 7-amino acid epitope present on the ECII of the receptor is also present as a conserved highly immunogenetic sequence present in the viral coat proteins, VP-1 and VP-2, of Parvovirus B19, a common human pathogen Citation[98]. Monoclonal antibodies from a Parvovirus B19-infected individual that are directed to the highly immunogenetic epitope on the viral coat proteins will also activate the receptor because of their interaction with the homologous region on the second loop of the AT1R Citation[49]. It is also possible that placental ischemia and the resulting tissue damage and inflammation may create a favorable setting for autoimmunity Citation[69]. The fact that the placenta has the highest tissue level of AT1R may also contribute to enhanced immunogenicity in a setting of placental ischemia. It is well known that a chronic inflammatory response activates the adaptive arm of the immune system and may create an environment that is permissive for autoimmunity.
Mechanism of autoantibody-mediated GPCR activation
GPCRs were long considered to function as monomers. However, a large body of biochemical evidence published in recent years clearly indicates that GPCRs form homodimers, heterodimers, and possibly higher order oligomeric structures Citation[100–102]. Agonist-induced dimerization has been shown for a number of GPCRs. Because receptor-activating antibodies are bivalent, it is possible that they exert their agonistic effect by cross-linking and thereby stabilizing receptor homodimers . Receptor-activating autoantibodies may serve as valuable investigative tools to understand the mechanism of agonist-induced receptor activation. A detailed analysis of antibody-receptor interaction has been carried out by Sanders and colleagues Citation[15] who prepared a complex consisting of the Fab fragment of the Graves’ monoclonal autoantibody (M22) and the extracellular domain of the TSHR (specifically, the first 260 amino acids of the LRD). X-ray diffraction analysis revealed that the antibody binds extensively to the concave surface of the LRD in a way that is believed to be nearly identical to that of the natural ligand. Thus, the structural analysis of autoantibody binding to the external domain of TSHR suggests that antibody-mediated receptor activation is accomplished in a very similar way to the natural ligand, TSH. However, the mechanism by which TSH or autoantibody binding to the LRD initiates transmission of a signal to the B-subunit (the transmembrane domain) followed by heterotrimeric G-protein activation is not known.
One of the most well-studied GPCRs is the angiotensin receptor AT1. Abdalla et al. have shown that the AT1 receptor forms heterodimers with the bradykinin B2 receptor and that heterodimer formation is associated with enhanced Ang II responsiveness Citation[103,104]. A thorough knowledge of the mechanism of antibody-induced receptor activation may provide further useful insights for the development of therapeutic strategies to block the action of the antibodies, thereby reducing the detrimental effects of excessive receptor activation.
Expert commentary & five-year view
Cardiomyopathy and hypertensive disorders are life-threatening diseases with the highest morbidity and mortality, affecting billions worldwide. In most cases, the fundamental basis for the disease is unknown. Also lacking are generally useful presymptomatic screening tests to identify individuals at risk prior to the development of symptoms. As reviewed here, autoantibodies capable of activating the AT1, ETA and the norepinephrine receptors (α1 and β1) may be important contributors to cardiovascular disease . A growing body of evidence indicates that autoantibodies capable of activating these receptors exist in the circulation of patients with cardiovascular disease, that these autoantibodies contribute to disease pathophysiology and that antibody titers correlate to the severity of the disease. The production of these pathogenic autoantibodies most likely precedes the onset of clinical symptoms, a possibility that highlights the autoantibodies as valuable presymptomatic markers. In view of the considerable evidence that these autoantibodies contribute to disease, they are likely to be important therapeutic targets in the management of cardiovascular disease. In the coming years we can expect to see continued development of improved immunological and functional tests to detect and quantify these pathogenic autoantibodies, and continued development of therapeutic strategies to remove these pathogenic autoantibodies or inhibit their ability to activate their target receptors. It is encouraging to see initial promising results from these approaches for patients with antibody-mediated graft rejection/malignant hypertension Citation[82] and patients with refractory hypertension Citation[39].
Based on compelling evidence for the pathogenic contributions of receptor-activating autoantibodies and the promising clinical trials showing the potential benefits of therapeutic removal, we can expect to see well-controlled clinical studies in the future to evaluate these therapeutic approaches for the treatment of these newly recognized autoimmune conditions. The studies reviewed here support the rapidly emerging concept that certain cardiovascular diseases contain a significant autoimmune component associated with pathological actions of harmful GPCR-activating autoantibodies. Insight provided by the recent findings reviewed here immediately suggests novel antibody-based presymptomatic testing and therapeutic approaches based on antibody neutralization or removal. If cardiovascular diseases have a major autoimmune component this would represent a significant and relatively prominent addition to the growing list of recognized autoimmune conditions.
Key issues
• Autoimmune diseases represent a major medical burden that affects an estimated 5% of the US population.
• Diseases characterized by the presence of receptor-activating autoantibodies are rapidly emerging as a specific autoimmune disease category.
• The classic example is Graves’ hyperthyroidism, in which autoantibodies activate the thyroid-stimulating hormone receptor resulting in overproduction of thyroid hormones.
• Considerable evidence now exists that agonistic autoantibodies targeting the cardiac β1-adrenergic receptor are associated with dilated cardiomyopathy and contribute to disease pathology.
• Recent evidence indicates that autoantibodies capable of activating α1-adrenergic receptors contribute to refractory hypertension and cardiomyopathy.
• Preeclampsia, a serious hypertensive disease of pregnancy, is characterized by the presence of autoantibodies that activate the major angiotensin receptor (AT1). The introduction of these autoantibodies into pregnant mice produces clinical features of preeclampsia.
• AT1 receptor-activating autoantibodies are also associated with kidney transplant recipients suffering from severe vascular rejection and malignant hypertension.
• Therapeutic removal of pathogenic autoantibodies by plasmaphoresis or immunoabsorption has proven beneficial to patients with renal allograft rejection and patients with refractory hypertension.
• AT1 receptor-activating autoantibodies as well as autoantibodies that activate the endothelin-1 receptor (ETA) are commonly observed in individuals diagnosed with systemic sclerosis.
• Receptor-activating autoantibodies represent potential presymptomatic markers and therapeutic targets for the medical management of cardiovascular diseases in which receptor-activating autoantibodies are involved.
Financial & competing interests disclosure
Support for this work was provided by the National Institute of Health grants HL076558, HD34130 and RC4HD067977, American Heart Association Grant 10GRNT3760081, March of Dimes (6-FY06-323) and the Texas Higher Education Coordinating Board. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Related Research Data
References
- Davies TF, Ando T, Lin RY, Tomer Y, Latif R. Thyrotropin receptor-associated diseases: from adenomata to Graves disease. J. Clin. Invest.115(8), 1972–1983 (2005).
- Michalek K, Morshed SA, Latif R, Davies TF. TSH receptor autoantibodies. Autoimmun. Rev.9(2), 113–116 (2009).
- Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol.84(3), 223–243 (1997).
- Adams DD, Fastier FN, Howie JB, Kennedy TH, Kilpatrick JA, Stewart RD. Stimulation of the human thyroid by infusions of plasma containing LATS protector. J. Clin. Endocrinol. Metab.39(5), 826–832 (1974).
- Costagliola S, Rodien P, Many MC, Ludgate M, Vassart G. Genetic immunization against the human thyrotropin receptor causes thyroiditis and allows production of monoclonal antibodies recognizing the native receptor. J. Immunol.160(3), 1458–1465 (1998).
- Laurberg P, Bournaud C, Karmisholt J, Orgiazzi J. Management of Graves’ hyperthyroidism in pregnancy: focus on both maternal and foetal thyroid function, and caution against surgical thyroidectomy in pregnancy. Eur. J. Endocrinol./Eur. Federat. Endocrine Soc.160(1), 1–8 (2009).
- Smith BR, Hall R. Thyroid-stimulating immunoglobulins in Graves’ disease. Lancet2(7878), 427–431 (1974).
- Ajjan RA, Weetman AP. Techniques to quantify TSH receptor antibodies. Nat. Clin. Pract. Endocrinol. Metab.4(8), 461–468 (2008).
- Sanders J, Evans M, Premawardhana LD et al. Human monoclonal thyroid stimulating autoantibody. Lancet362(9378), 126–128 (2003).
- Smith BR, Bolton J, Young S et al. A new assay for thyrotropin receptor autoantibodies. Thyroid14(10), 830–835 (2004).
- Nakatake N, Sanders J, Richards T et al. Estimation of serum TSH receptor autoantibody concentration and affinity. Thyroid16(11), 1077–1084 (2006).
- El Fassi D, Banga JP, Gilbert JA, Padoa C, Hegedus L, Nielsen CH. Treatment of Graves’ disease with rituximab specifically reduces the production of thyroid stimulating autoantibodies. Clin. Immunol.130(3), 252–258 (2009).
- Sanders J, Jeffreys J, Depraetere H et al. Characteristics of a human monoclonal autoantibody to the thyrotropin receptor: sequence structure and function. Thyroid14(8), 560–570 (2004).
- Smith BR, Sanders J, Furmaniak J. TSH receptor antibodies. Thyroid17(10), 923–938 (2007).
- Sanders J, Chirgadze DY, Sanders P et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid17(5), 395–410 (2007).
- Costagliola S, Many MC, Denef JF, Pohlenz J, Refetoff S, Vassart G. Genetic immunization of outbred mice with thyrotropin receptor cDNA provides a model of Graves’ disease. J. Clin. Invest.105(6), 803–811 (2000).
- Nagayama Y, Kita-Furuyama M, Ando T et al. A novel murine model of Graves’ hyperthyroidism with intramuscular injection of adenovirus expressing the thyrotropin receptor. J. Immunol.168(6), 2789–2794 (2002).
- Borda E, Pascual J, Cossio P, De La Vega M, Arana R, Sterin-Borda L. A circulating IgG in Chagas’ disease which binds to β-adrenoceptors of myocardium and modulates their activity. Clin. Exp. Immunol.57(3), 679–686 (1984).
- Jahns R, Boivin V, Lohse MJ. β(1)-Adrenergic receptor function, autoimmunity, and pathogenesis of dilated cardiomyopathy. Trends Cardiovasc. Med.16(1), 20–24 (2006).
- Limas CJ, Goldenberg IF, Limas C. Autoantibodies against β-adrenoceptors in human idiopathic dilated cardiomyopathy. Circ. Res.64(1), 97–103 (1989).
- Magnusson Y, Marullo S, Hoyer S et al. Mapping of a functional autoimmune epitope on the β 1-adrenergic receptor in patients with idiopathic dilated cardiomyopathy. J. Clin. Invest.86(5), 1658–1663 (1990).
- Wallukat G, Morwinski M, Kowal K, Forster A, Boewer V, Wollenberger A. Autoantibodies against the β-adrenergic receptor in human myocarditis and dilated cardiomyopathy: β-adrenergic agonism without desensitization. Eur. Heart. J.12(Suppl. D), 178–181 (1991).
- Magnusson Y, Wallukat G, Waagstein F, Hjalmarson A, Hoebeke J. Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the β 1-adrenoceptor with positive chronotropic effect. Circulation89(6), 2760–2767 (1994).
- Magnusson Y, Hjalmarson A, Hoebeke J. β 1-adrenoceptor autoimmunity in cardiomyopathy. Int. J. Cardiol.54(2), 137–141 (1996).
- Mobini R, Fu M, Wallukat G, Magnusson Y, Hjalmarson A, Hoebeke J. A monoclonal antibody directed against an autoimmune epitope on the human β 1-adrenergic receptor recognized in idiopathic dilated cardiomyopathy. Hybridoma19(2), 135–142 (2000).
- Nikolaev VO, Boivin V, Stork S et al. A novel fluorescence method for the rapid detection of functional β 1-adrenergic receptor autoantibodies in heart failure. J. Am. Coll. Cardiol.50(5), 423–431 (2007).
- Wallukat G, Nissen E, Morwinski R, Muller J. Autoantibodies against the β- and muscarinic receptors in cardiomyopathy. Herz25(3), 261–266 (2000).
- Munoz-Saravia SG, Haberland A, Wallukat G, Schimke I. Chronic Chagas’ heart disease: a disease on its way to becoming a worldwide health problem: epidemiology, etiopathology, treatment, pathogenesis and laboratory medicine. Heart Fail. Rev. DOI: 10.1007/s10741-010-9211-5 (2010) (Epub ahead of print).
- Wallukat G, Munoz Saravia SG, Haberland A et al. Distinct patterns of autoantibodies against G-protein-coupled receptors in Chagas’ cardiomyopathy and megacolon. Their potential impact for early risk assessment in asymptomatic Chagas’ patients. J. Am. Coll. Cardiol.55(5), 463–468 (2010).
- Jahns R, Boivin V, Schwarzbach V, Ertl G, Lohse MJ. Pathological autoantibodies in cardiomyopathy. Autoimmunity41(6), 454–461 (2008).
- Ferrari I, Levin MJ, Wallukat G et al. Molecular mimicry between the immunodominant ribosomal protein P0 of Trypanosoma cruzi and a functional epitope on the human β 1-adrenergic receptor. J. Exp. Med.182(1), 59–65 (1995).
- Wallukat G, Wollenberger A. Effects of the serum γ globulin fraction of patients with allergic asthma and dilated cardiomyopathy on chronotropic β adrenoceptor function in cultured neonatal rat heart myocytes. Biomedica Biochimica Acta46(8–9), S634–S639 (1987).
- Jahns R, Boivin V, Hein L et al. Direct evidence for a β 1-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy. J. Clin. Invest.113(10), 1419–1429 (2004).
- Jahns R, Boivin V, Lohse MJ. β 1-adrenergic receptor-directed autoimmunity as a cause of dilated cardiomyopathy in rats. Int. J. Cardiol.112(1), 7–14 (2006).
- Christ T, Wettwer E, Dobrev D et al. Autoantibodies against the β 1 adrenoceptor from patients with dilated cardiomyopathy prolong action potential duration and enhance contractility in isolated cardiomyocytes. J. Mol. Cell. Cardiol.33(8), 1515–1525 (2001).
- Deubner N, Berliner D, Schlipp A et al. Cardiac β 1-adrenoceptor autoantibodies in human heart disease: rationale and design of the Etiology, Titre-Course, and Survival (ETiCS) Study. Eur. J. Heart Fail.12(7), 753–762 (2010).
- Fu ML, Herlitz H, Wallukat G et al. Functional autoimmune epitope on α 1-adrenergic receptors in patients with malignant hypertension. Lancet344(8938), 1660–1663 (1994).
- Luther HP, Homuth V, Wallukat G. α 1-adrenergic receptor antibodies in patients with primary hypertension. Hypertension29(2), 678–682 (1997).
- Wenzel K, Haase H, Wallukat G et al. Potential relevance of α(1)-adrenergic receptor autoantibodies in refractory hypertension. PLoS ONE3(11), e3742 (2008).
- Yan L, Xu Y, Yao H et al. The effects of autoantibodies against the second extracellular loop of α(1)-adrenoceptor on vasoconstriction. Basic Res. Cardiol.104(5), 581–589 (2009).
- Wenzel K, Wallukat G, Qadri F et al. α 1A-adrenergic receptor-directed autoimmunity induces left ventricular damage and diastolic dysfunction in rats. PLoS ONE5(2), e9409 (2010).
- Karczewski P, Haase H, Hempel P, Bimmler M. Agonistic antibody to the α 1-adrenergic receptor mobilizes intracellular calcium and induces phosphorylation of a cardiac 15-kDa protein. Mol. Cell. Biochem.333(1–2), 233–242 (2009).
- Young BC, Levine RJ, Karumanchi SA. Pathogenesis of preeclampsia. Ann. Rev. Pathol.5, 173–192 (2010).
- Roberts JM, Cooper DW. Pathogenesis and genetics of pre-eclampsia. Lancet357, 53–56 (2001).
- Xia Y, Zhou CC, Ramin SM, Kellems RE. Angiotensin receptors, autoimmunity, and preeclampsia. J. Immunol.179(6), 3391–3395 (2007).
- Lamarca BD, Gilbert J, Granger JP. Recent progress toward the understanding of the pathophysiology of hypertension during preeclampsia. Hypertension51(4), 982–988 (2008).
- Xia Y, Kellems RE. Is preeclampsia an autoimmune disease? Clin. Immunol.133(1), 1–12 (2009).
- Wenzel K, Rajakumar A, Haase H et al. Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension58(1), 77–84 (2011).
- Herse F, Verlohren S, Wenzel K et al. Prevalence of agonistic autoantibodies against the angiotensin II type 1 receptor and soluble fms-like tyrosine kinase 1 in a gestational age-matched case study. Hypertension53(2), 393–398 (2009).
- Wallukat G, Homuth V, Fischer T et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Invest.103(7), 945–952 (1999).
- Dechend R, Viedt C, Muller DN et al. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation107(12), 1632–1639 (2003).
- Dechend R, Homuth V, Wallukat G et al. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation101(20), 2382–2387 (2000).
- Bobst SM, Day MC, Gilstrap LC 3rd, Xia Y, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human mesangial cells and induce interleukin-6 and plasminogen activator inhibitor-1 secretion. Am. J. Hypertens.18(3), 330–336 (2005).
- Xia Y, Wen H, Bobst S, Day MC, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human trophoblast cells. J. Soc. Gynecol. Invest.10(2), 82–93 (2003).
- Zhou CC, Ahmad S, Mi T et al. Autoantibody from women with preeclampsia induces soluble Fms-like tyrosine kinase-1 production via angiotensin type 1 receptor and calcineurin/nuclear factor of activated T-cells signaling. Hypertension51(4), 1010–1019 (2008).
- Zhou CC, Irani RA, Zhang Y et al. Angiotensin receptor agonistic autoantibody-mediated tumor necrosis factor-α induction contributes to increased soluble endoglin production in preeclampsia. Circulation121(3), 436–444 (2010).
- Yang X, Wang F, Chang H et al. Autoantibody against AT1 receptor from preeclamptic patients induces vasoconstriction through angiotensin receptor activation. J. Hypertens.26(8), 1629–1635 (2008).
- Zhou CC, Zhang Y, Irani RA et al. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat. Med.14(8), 855–862 (2008).
- Siddiqui AH, Irani RA, Blackwell SC, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody is highly prevalent in preeclampsia: correlation with disease severity. Hypertension55(2), 386–393 (2010).
- Kaufmann P, Black S, Huppertz B. Endovascular trophoblast invasion: implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol. Reproduct.69(1), 1–7 (2003).
- Barker DJ. In utero programming of chronic disease. Clin. Sci. (Lond.)95(2), 115–128 (1998).
- Godfrey KM, Barker DJ. Fetal nutrition and adult disease. Am. J. Clin. Nutrit.71(Suppl. 5), S1344–S1352 (2000).
- Baum M, Ortiz L, Quan A. Fetal origins of cardiovascular disease. Curr. Opin. Pediat.15(2), 166–170 (2003).
- Irani RA, Zhang Y, Blackwell SC et al. The detrimental role of angiotensin receptor agonistic autoantibodies in intrauterine growth restriction seen in preeclampsia. J. Exp. Med.206(12), 2809–2822 (2009).
- Gadonski G, Lamarca BB, Sullivan E, Bennett W, Chandler D, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of interleukin 6. Hypertension48(4), 711–716 (2006).
- Gilbert JS, Ryan MJ, Lamarca BB, Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension during preeclampsia: linking placental ischemia with endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol.294(2), H541–H550 (2008).
- Parrish MR, Murphy SR, Rutland S et al. The effect of immune factors, tumor necrosis factor-α, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am. J. Hypertens.23(8), 911–916 (2010).
- Li Z, Zhang Y, Ying Ma J et al. Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension50(4), 686–692 (2007).
- LaMarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor α in pregnant rats. Hypertension52(6), 1168–1172 (2008).
- Lamarca B, Parrish M, Ray LF et al. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension54(4), 905–909 (2009).
- Lamarca B, Wallace K, Herse F et al. Hypertension in response to placental ischemia during pregnancy: role of B lymphocytes. Hypertension57(4), 865–871 (2011).
- Wallace K, Richards S, Dhillon P et al. CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension57(5), 949–955 (2011).
- Dechend R, Gratze P, Wallukat G et al. Agonistic autoantibodies to the AT1 receptor in a transgenic rat model of preeclampsia. Hypertension45(4), 742–746 (2005).
- Zhang SL, Du YH, Wang J et al. Endothelial dysfunction induced by antibodies against angiotensin AT1 receptor in immunized rats. Acta Pharmacol. Sin.31(10), 1381–1388 (2010).
- Irani RA, Zhang Y, Zhou CC et al. Autoantibody-mediated angiotensin receptor activation contributes to preeclampsia through tumor necrosis factor-α signaling. Hypertension55(5), 1246–1253 (2010).
- Zhou CC, Irani RA, Dai Y et al. Autoantibody-mediated IL-6-dependent endothelin-1 elevation underlies pathogenesis in a mouse model of preeclampsia. J. Immunol.186(10), 6024–6034 (2011).
- Zhou CC, Irani RA, Zhang Y et al. Angiotensin receptor agonistic autoantibody-mediated tumor necrosis factor-α induction contributes to increased soluble endoglin production in preeclampsia. Circulation121(3), 436–444 (2010).
- Parrish MR, Wallace K, Tam Tam KB et al. Hypertension in response to AT1-AA: Role of reactive oxygen species in pregnancy-induced hypertension. Am. J. Hypertens.24(7), 835–840 (2011).
- Parrish MR, Ryan MJ, Glover P et al. Angiotensin II type 1 autoantibody induced hypertension during pregnancy is associated with renal endothelial dysfunction. Gender Med.8(3), 184–188.
- LaMarca B, Wallace K, Granger J. Role of angiotensin II type I receptor agonistic autoantibodies (AT1-AA) in preeclampsia. Curr. Opin. Pharmacol.11(2), 175–179 (2011).
- Dragun D, Brasen JH, Schonemann C et al. Patients with steroid refractory acute vascular rejection develop agonistic antibodies targeting angiotensin II type 1 receptor. Transplant. Proc.35(6), 2104–2105 (2003).
- Dragun D, Muller DN, Brasen JH et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N. Engl. J. Med.352(6), 558–569 (2005).
- Dragun D. Agonistic antibody-triggered stimulation of Angiotensin II type 1 receptor and renal allograft vascular pathology. Nephrol. Dial. Transplant.22(7), 1819–1822 (2007).
- Fu ML, Herlitz H, Schulze W et al. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J. Hypertens.18(7), 945–953 (2000).
- Wei F, Jia XJ, Yu SQ et al. Candesartan versus imidapril in hypertension: a randomised study to assess effects of anti-AT1 receptor autoantibodies. Heart97(6), 479–484 (2011).
- Sun YX, Zhang HY, Wei YM, Zhu F, Wang M, Liao YH. The mechanism of signal transduction during vascular smooth muscle cell proliferation induced by autoantibodies against angiotensin AT1 receptor from hypertension. Chinese Med. J.121(1), 43–48 (2008).
- Liao YH, Wei YM, Wang M, Wang ZH, Yuan HT, Cheng LX. Autoantibodies against AT1-receptor and α1-adrenergic receptor in patients with hypertension. Hypertens. Res.25(4), 641–646 (2002).
- Dragun D. The role of angiotensin II type 1 receptor-activating antibodies in renal allograft vascular rejection. Pediat. Nephrol. (Berlin, Germany)22(7), 911–914 (2007).
- Reinsmoen NL, Lai CH, Heidecke H et al. Anti-angiotensin type 1 receptor antibodies associated with antibody mediated rejection in donor HLA antibody negative patients. Transplantation90(12), 1473–1477 (2010).
- Riemekasten G, Philippe A, Nather M et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann. Rheum. Dis.70(3), 530–536 (2010).
- Walllukat GDM, Muller J, Bartel S, Schulze W, Hetzer R. Agonistic autoantibodies against the endothelin 1 ETA- and α 1-adrenergic receptor in the sera of patients with idiopathic pulmonary arterial hypertension. Circulation116(II), 503 (2007).
- Dandel MWG, Englert A, Lehmkul HB, Hetzer R. Potential pathogenetic involvement of agonistic autoantibodies against α-1 adrenergic and endothelin-1 receptors in pulmonary arterial hypertension. Circulation118, S1072 (2008).
- Dandel MWG, Englert A, Lehmkul HB, Hetzer R. Agonistic autoantibodies against α-1 adrenergic and endothelin-1 receptors in sera of patients with pulmonary arterial hypertension: potential involvement in the pathogenesis of the disease. J. Heart Lung Transplant.28(2), S249 (2009).
- Dragun D, Philippe A, Catar R, Hegner B. Autoimmune mediated G-protein receptor activation in cardiovascular and renal pathologies. Thrombo. Haemost.101(4), 643–648 (2009).
- Walther T, Wallukat G, Jank A et al. Angiotensin II type 1 receptor agonistic antibodies reflect fundamental alterations in the uteroplacental vasculature. Hypertension46(6), 1275–1279 (2005).
- Chen CR, Pichurin P, Nagayama Y, Latrofa F, Rapoport B, Mclachlan SM. The thyrotropin receptor autoantigen in Graves disease is the culprit as well as the victim. J. Clin. Invest.111(12), 1897–1904 (2003).
- Mizutori Y, Chen CR, Latrofa F, Mclachlan SM, Rapoport B. Evidence that shed thyrotropin receptor A subunits drive affinity maturation of autoantibodies causing Graves’ disease. J. Clin. Endocrinol. Metab.94(3), 927–935 (2009).
- Gigler A, Dorsch S, Hemauer A et al. Generation of neutralizing human monoclonal antibodies against parvovirus B19 proteins. J. Virol.73(3), 1974–1979 (1999).
- Stepan H, Wallukat G, Schultheiss HP, Faber R, Walther T. Is parvovirus B19 the cause for autoimmunity against the angiotensin II type receptor? J. Reprod. Immunol.73(2), 130–134 (2007).
- Bai M. Dimerization of G-protein-coupled receptors: roles in signal transduction. Cell. Signalling16(2), 175–186 (2004).
- Breitwieser GE. G protein-coupled receptor oligomerization: implications for G protein activation and cell signaling. Cir. Res.94(1), 17–27 (2004).
- Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Reports5(1), 30–34 (2004).
- Abdalla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature407(6800), 94–98 (2000).
- Abdalla S, Lother H, El Massiery A, Quitterer U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat. Med.7(9), 1003–1009 (2001).
- Szkudlinski MW, Fremont V, Ronin C, Weintraub BD. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol. Rev.82(2), 473–502 (2002).