
Abstract
Common variable immunodeficiency (CVID) is the most common symptomatic primary immunodeficiency in adults. As symptoms of CVID are usually heterogeneous and unspecific, diagnosis and follow-up of CVID can be challenging. In light of this, a broad review of advances in management and treatment of CVID is performed here in order to reach a distinct protocol. However, it should be noted that owing to the nature of the disease, it can only be treated symptomatically but not cured. There is little evidence to guide appropriate or universal guidelines to improve the current status of management of the disease. The most satisfactory treatments of CVID could be achieved by the use of immunoglobulin replacement, antibiotics, immunosuppressants and hematopoietic stem cell transplantation. This review is written based on the importance of clinical surveillance of asymptomatic CVID cases and early recognition of different clinical complications. Moreover, for each complication, appropriate interventions for improving outcomes are mentioned.
Medscape: Continuing Medical Education Online
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Expert Reviews Ltd. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 70% minimum passing score and complete the evaluation at www.medscape.org/journal/expertimmunology; (4) view/print certificate.
Release date: 30 May 2013; Expiration date: 30 May 2014
Learning objectives
Upon completion of this activity, participants will be able to:
• Analyze the clinical presentation of CVID
• Assess the practice of exogenous IgG administration to patients with CVID
• Evaluate pulmonary complications of CVID and their management
• Evaluate other potential complications of CVID
Financial & competing interests disclosure
EDITOR
Elisa Manzotti
Publisher, Future Science Group, London, UK.
Disclosure: Elisa Manzotti has disclosed no relevant financial relationships.
CME AUTHOR
Charles P Vega, MD
Associate Professor and Residency Director, Department of Family Medicine, University of California, Irvine.
Disclosure: Charles P Vega, MD, has disclosed no relevant financial relationships
AUTHORS AND CREDENTIALS
Hassan Abolhassani
Research Center for Immunodeficiencies, Pediatrics Center of Excellence, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
Disclosure: Hassan Abolhassani has disclosed no relevant financial relationships.
Babak Torabi Sagvand
Research Center for Immunodeficiencies, Pediatrics Center of Excellence, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
Disclosure: Babak Torabi Sagvand has disclosed no relevant financial relationships.
Tahaamin Shokuhfar
Research Center for Immunodeficiencies, Pediatrics Center of Excellence, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
Disclosure: Tahaamin Shokuhfar has disclosed no relevant financial relationships.
Babak Mirminachi
Research Center for Immunodeficiencies, Pediatrics Center of Excellence, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
Disclosure: Babak Mirminachi has disclosed no relevant financial relationships.
Nima Rezaei
Research Center for Immunodeficiencies, Pediatrics Center of Excellence, Children’s Medical Center, Tehran University of Medical Sciences; Molecular Immunology Research Center and Department of Immunology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran.
Disclosure: Nima Rezaei has disclosed no relevant financial relationships.
Asghar Aghamohammadi
Research Center for Immunodeficiencies, Pediatrics Center of Excellence, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
Disclosure: Asghar Aghamohammadi has disclosed no relevant financial relationships.
CVID: Common variable immunodeficiency; HIGM: Hyper IgM syndrome; ICOS: Inducible T-cell costimulator; SCID: Severe combined immunodeficiency; XLA: X-linked agammaglobulinemia; XLP: X-linked lymphoproliferative disease.
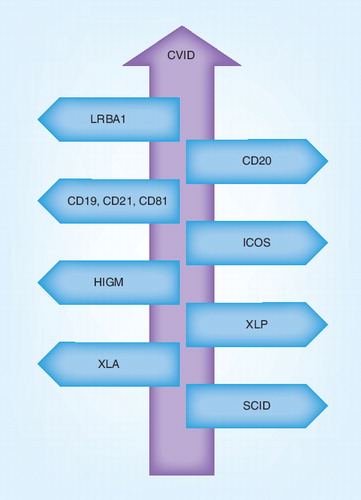
Common variable immune deficiency (CVID) as a heterogeneous group of primary immune deficiencies is characterized by insufficient serum levels of immunoglobulins (Igs), reduced response to specific antigens and higher incidence of repeated infections Citation[1–4]. Autoimmune disorders, lymphoproliferative disorders and gastric complications also have an increased incidence in patients with CVID Citation[5,6]. The prevalence of this predominantly antibody deficiency should not be thought of as being rare (1:10,000–50,000) and CVID is the most common symptomatic human primary immunodeficiency Citation[7–9]. The onset (early or late) and clinical manifestations (distinct phenotypes) of the disease have heterogenous presentations Citation[10–12]. This variability in clinical phenotype and late onset of disease may lead to delay in CVID diagnosis Citation[13,14].
The molecular basis, both immunologically and genetically, of CVID is still unclear despite huge amounts of evaluation in this field since 1953 Citation[15]. However, during the last 10 years, 10–15% of the patients with a history of CVID manifestations are classified in other primary immunodeficiency categories because of specific identified gene mutations including ICOS, CD19, CD20, CD81, CD21 and LRBA1 Citation[16–23]. Decreased serum level of IgG, IgA and/or IgM (at least 2 standard deviations below the mean for the age group), hyporesponsiveness to specific antigens, minimum age of 4 years, absence of lymphoid malignancy during the first 2 years of diagnosis and genetic exclusion of other known etiologies for hypogammaglobulinemia are considered as criteria for diagnosis of CVID Citation[24–26]. Furthermore, approximately 2% of CVID patients may represent clinical or laboratory features that suggest a known severe combined primary immunodeficiency (e.g., opportunistic fungal or viral infections, very low numbers of T cells and/or monocytes). Although this phenomenon is rare, this is important because such patients may need stem cell grafts Citation[27].
Heterogeneity in CVID refers to variability in immunological and genetic defects and diversity in clinical symptoms described in this group of patients Citation[19,28,29]. The main immunological defect is failure of B-cell Ig production, although abnormalities have been described in all other components of the immune system Citation[30,31]. Only some CVID cases are due to monogenic Mendelian diseases and homozygous mutations in that some genes have been reported in some CVID families Citation[17,18,32].
The clinical spectrum of CVID is broad. The main clinical manifestations are recurrent infections occurring in the respiratory tract, GI tract, skin and soft tissues Citation[4,33,34]. However, inflammatory complications occur in varying proportions and include autoimmunity, chronic lung disease, bronchiectasis, gastrointestinal (GI) disease with or without malabsorption, systemic or localized granulomatous disease, liver disease, splenomegaly, lymphadenopathy with or without lymphoma and other malignancies Citation[26,35]. By using data of clinical manifestations and complications from 334 CVID patients from seven European centers, five different clinical phenotypes were considered for these patients Citation[13]. In the revised phenotyping criteria, lymphoid malignancies were excluded Citation[36].
Over the last few years, advances have been made in the management of CVID, improving outcomes in the patients; these include Ig replacement, antibiotics for treatment and prevention of infections and appropriate therapy for noninfectious complications Citation[37].
IgG replacement is the mainstay of treatment of CVID Citation[35] and it has been shown that long-term Ig replacement therapy for CVID has reduced the rate of infections and their long-term complications Citation[38–41]. However, despite the reduction in the rate of bacterial infections by use of IgG replacement in CVID patients, these patients are still more susceptible to complications because of a dysregulated immune response that attracts the caregiver’s attention. Here, the authors review the recent advances in CVID, specifically the clinical features, and focus on the management and treatment of CVID. Other specific classifications based on immunological property of CVID cases (including Freiburg, Paris, EUROclass and severe T-cell defect classification) exist, however their importance for clinical management are under evaluation Citation[42–46]. Moreover, the authors review the recent advances on the management and treatment of CVID.
Clinical manifestations of CVID
The clinical manifestations of CVID constitute six major categories including: infections, pulmonary complications, granulomatous or polyclonal lymphocytic infiltrative diseases, autoimmunity, GI diseases and neoplasias . These can be established in different periods of life, from childhood to late adulthood, with a bimodal age distribution, demonstrating two peaks between 1 and 5 years and 18 and 25 years Citation[3,4,47].
Management of infectious complications
Virtually all CVID patients encounter chronic or recurrent infections, particularly sinusitis, otitis, bronchitis and pneumonia Citation[47–49]. Approximately 90% of CVID patients have encountered at least one episode of chronic sinusitis and 70% have suffered recurrent otitis media before diagnosis Citation[50–52]. Previously, there had been a history of at least one episode of pneumonia before diagnosis in 75–85% of the CVID patients as well as multiple episodes in many others Citation[38,41]. The majority of morbidities and mortalities of CVID is due to long-term sequelae of recurrent respiratory tract infections including chronic sinusitis, hearing loss (due to tympanic membrane perforation) and bronchiectasis Citation[4]. Encapsulated (Haemophylus influenzae, Streptococcus pneumoniae) or atypical (Mycoplasma spp.) bacteria are the major causative agents for recurrent infections of both the upper and the lower respiratory tract Citation[4,35,53]. Failure of antigen-specific IgG production, which increases susceptibility to encapsulated bacteria, seems to be responsible for recurrent rhinoviral infections in CVID Citation[51].
The GI tract is the second organ that is involved in infections in 10–40% of the CVID cases. Various pathogens can cause GI infections in CVID patients including: Giardia lamblia, Campylobacter jejuni, Salmonella spp., Cryptosporidium parvum, CMV, Clostridium difficile, Helicobacter pylori, HBV, HCV and so on. Giardiasis is the most prevalent GI infection, especially in those with undetectable serum IgA levels Citation[54]. H. pylori is an important pathogen in CVID, resulting in chronic active gastritis involving antrum and corpus, achlorhydria, gastric adenocarcinoma and gastric lymphoma Citation[55,56].
The CNS (meningoencephalitis is especially caused by Enterovirus spp.), joints, bones (especially due to Mycoplasma spp.), skin and eyes are also affected by CVID and their involvement might be the first and sole presentation of CVID Citation[57]. Fibrotic bladder can develop due to recurrent urinary tract infections due to Ureaplasma urealyticum Citation[25,36].
Viral hepatitis (especially HCV transmitted by intravenous Ig [IVIG] administration) and severe Herpes zoster infection have been reported in large numbers of patients Citation[4]. Because of recent developments in Ig preparation including careful selection of donors, plasma antibody screening and effective procedures of viral inactivation, the rate of these infections are now very rare in the western world Citation[58]. CMV is involved in many inflammatory complications of a selected group of CVID patients with special immune cell phenotypes Citation[59]. Enteroviral infection is also currently rare in western CVID patients, however this microorganism being maintained probably due to lack of high-dose treatment and may lead to meningoencephalitis with poor prognosis Citation[60]. The minority of these patients are prone to meningoencephalitis, which usually has a poor prognosis Citation[61]. Human parechovirus type 1 and norovirus were also reported in CVID patients that had severe enteropathy Citation[62].
Prevention
Lifelong Ig replacement therapy (injection of human antibodies harvested from plasma donations) is the mainstay of the therapeutic approach for stopping the cycle of recurrent infections in CVID patients. Both intravenous (3–12% IVIG) and subcutaneous (10–20% subcutaneous Ig [SCIG]) routes provide sufficient amounts of Ig Citation[58,63]. IVIG is used more commonly than SCIG and reduces both the rate of acute and chronic infections and their long-term secondary medical conditions in CVID Citation[38,40,64]. Although the purpose of Ig replacement therapy is to prevent infections, the amounts of administered Ig and individualized dosing can vary depending on baseline level of IgG and presence of chronic lung or GI damage Citation[65]. Furthermore, the optimum trough level of IgG is not universal and clinical response may be a better indicator for dose adjustments in CVID cases Citation[66–68]. Based on this goal, universal IVIG routine protocols recommend typically starting the dose at 400–600 mg/kg every 3–4 weeks, while SCIG is usually started by dosage at 100–200 mg/kg followed by 160 mg/kg every week. In infants and young children, 5–7 ml of SCIG per site is well tolerated; in adults, the volume per site is generally 15–20 ml Citation[69–72].
Although long-term IVIG infusion is an effective treatment for prevention of recurrent infections in CVID patients, it can be complicated by systemic adverse reactions. These side effects may occur up to 72 h after the infusion Citation[73,74].
The majority of IVIG side effects are mild, transient and self-limited and do not require discontinuation of therapy Citation[58]. Predisposing factors responsible for adverse reactions include infection Citation[75], rate of infusion, other comorbidities or patient-related factors such as age (≥65 years), cardiovascular impairment, renal dysfunction, thromboembolic risk and the presence of diabetes mellitus, pre-existing renal disease, hypovolemia, sepsis, anti-IgA antibodies and increase of interval since last infusion Citation[76]. One study showed that side effects of IVIG were not significantly correlated with pregnancy Citation[77]. There are different types of Ig products regarding the concentration of antibodies and other plasma proteins such as sodium content, sugar content, osmolarity and IgA content. The selection of Ig product must be individualized based on clinical condition of the patient and Ig product-related factors. For patients with congestive heart failure and elderly patients, Ig products with 10% concentration and lower sodium content are more suitable Citation[78].
Monitoring for adverse reactions to IVIG and SCIG must be performed during therapy. Depending on the type of adverse reactions, different managements should be considered.
A very important issue to consider is determination of patients with anti-IgA. Some tests that detect IgG anti-IgA seem to be helpful in predicting adverse reactions. By contrast, some authors disagree with the significant importance of anti-IgA antibodies and they consider it as a rare problem in CVID. Furthermore, many expert centers do not measure these antibodies because there is little correlation between levels and reactions. However, low-IgA preparations or SCIG should be used in CVID cases who generate anti-IgA antibodies against routine IVIG. After prevention of all predisposing factors for adverse reactions, premedication including paracetamol (500–1000 mg), oral diphenhydramine (25–50 mg), corticosteroids/acetaminophen/anti-inflammatory drugs should be considered prior to treatment for patients who experience adverse reaction following IVIG infusions. If persistent headache symptoms are observed, the patient can switch to SCIG to eliminate the problem and no further premedication is needed Citation[79,80]. Despite this idea, many physicians do not agree with this strategy and they manage headache critically Citation[76].
Most patients tolerate IVIG and SCIG well if administered by an expert nurse. Avoiding infusion during active infection and changing the preparation in those rare patients with repeated reactions usually solves the problem. Moreover, hydration of patient (before, after and during treatment), preparation of Ig product before infusion and using a heating pad or warm blanket because of chilling or local swelling are other strategies to facilitate intravenous replacement therapy Citation[81–83]. Besides preference of SCIG in patients with major adverse events compared with IVIG, this type of replacement reduces fluctuations in serum IgG concentrations, does not require venous access, prepares rapid infusions, decreases risk of fluid overload or hyperosmolarity, is prescribed home-based self-administration requiring minimal skills, improves quality of life and saves travel time, reduces utilization of the healthcare system and eliminates missing work or school. However, local reactions to SCIG are unacceptable for patients with general edema or lack of subcutaneous tissue. Discomfort associated with the needle stick of SCIG can be minimized with local anesthetic creams Citation[58,71].
The second line in prevention of infection in CVID patients is antibiotic prophylaxis, especially in the cases with bronchiectasis, frequent infections (generally more than three per year) or disruptive infections (hospital admission, prolonged period off work, secondary complications such as empyema) Citation[84]. However, this effect of prophylactic antibiotics in CVID patients has not been rigorously assessed Citation[85]. Previous microbiology results, serial sputum testing and antibiotic sensitivity of cultured organisms determine the choice of antimicrobial prophylaxis Citation[86]. Daily use of trimethoprim– sulfamethoxazole or macrolides, which provide substantial anti-inflammatory effects, provide more benefit than much greater doses of Ig therapy in patients with persistent lung diseases Citation[14,87]. Other regimes for prophylaxis consist of azithromycin 250 mg three times/week, cotrimoxazole 960 mg three times/week, amoxicillin 500 mg two times/day and ciprofloxacin 250 mg two times/day Citation[88–90]. By contrast, some authors suggest that the use of prophylactic antibiotics should be avoided because of an increased risk of infection with fungi or other resistant organisms. However, resistant organisms can be treated if they arise by changing of antibiotics according to an algorithm of alternative antibiotics Citation[87].
Immunization by polysaccharide vaccines may be effective in selected CVID patients with normal class-switched memory B cells. Activated vaccine (MMR and varicella) may be neutralized by antibodies of IVIG and are not recommended in CVID. In contrast, antibody against seasonal influenza virus is not presented in IVIG, therefore yearly recommendation for inactivated influenza vaccine administration is acceptable. Moreover, for indirect protection, close-contact relatives of patients should be vaccinated against influenza Citation[91,92].
Follow-up
Pay attention to educate patients (and parents of children) about alarming signs of infections and the necessity of holding sputum pots themselves before starting antibiotics which is the main step for early detection of infections. Beside patients, raising the awareness of medical and social caregivers for appropriate approach to recurrent infection is critical. Clinical visits and physical examination for early detection of infections in CVID cases that warrant immediate treatment should be carried out every 3–6 months. Sputum monitoring and analysis must be performed for all cases with productive cough. Chronic viral infection is often very difficult to diagnose without a sophisticated virology laboratory. Although with current legislation on the control of Ig products and viral inactivation, no case of viral transmission by Ig administration has been reported since 2000, the HCV viral genome (HCV RNA) must be checked for regularly in all patients already receiving IVIG. Very recently, detection of κ-deleting recombination circles provides a tool for neonatal screening of B-cell formation defects before presentation of infections, but the sensitivity and specificity of this modality should be evaluated in CVID cases Citation[93,94].
Treatment
Early treatment of infections at the first signs and symptoms should be considered an integral part of the treatment of CVID to prevent secondary structural damage. In each type of infection, samples must be collected before oral or intravenous antibiotic therapy if possible. Although culture and sensitivity results help clinicians choose the most effective drugs, this should not lead to delayed empiric therapy. The effective empiric therapy for sinopulmonary infections in patients not taking prophylactic drugs (amoxicillin, macrolide or levofloxacin) differ from cases taking prophylactic drugs (amoxicillin clavulanate, macrolide or ciprofloxacin Citation[90]). Initial treatment in GI microorganisms, the second most important group of infections, is determined on the basis of culture results and biopsy findings and usually includes antibiotics, restoration of nutrients and rehydration.
The use of antiviral agents, such as gancliclovir, pleconaril or ribavirin, appears to be safe and sometimes effective but should be evaluated in controlled clinical trials Citation[60,95].
It should be noted that active infection plays a role in increasing the risk of adverse reaction to IVIG, therefore combination of antibiotic therapy and Ig replacement must be planned in these cases. A prolonged course of treatment should be considered in cases with relapse of infections. Because of the nature of the disease, resistant common organisms and unusual organisms (Pseudomonas spp., Mycobacterium tuberculosis and opportunistic infections) should be reviewed when treatment fails.
Management of pulmonary complications
As the most frequent manifestation of CVID, pulmonary diseases are present in most patients at the time of diagnosis and they significantly increase morbidity and mortality in CVID patients Citation[47,51]. Recurrent pulmonary infections with pyogenic bacteria among CVID patients can culminate in permanent pulmonary disorders such as air-trapping, ground-glass attenuation, parenchymal opacification, reticular fibrosis, bronchial wall thickening, atelectasis and bronchiectasis (obstructive and restrictive) Citation[48,51]. These complications can develop in various periods of life but are slightly milder among pediatric patients than in adult cases Citation[51]. Of the aforementioned complications, bronchiectasis subsequent to recurrent pneumonia is more prominent and develops in approximately a third of patients Citation[4,96]. Despite regular and adequate treatment in response to the immune dysregulation that occurs in CVID, affected patients may develop inflammatory diseases, which could result in more pulmonary complications, especially bronchiectasis, in this group of patients Citation[47,97].
Prevention
Prophylaxis and treatment of upper and lower respiratory tract infections with greater doses of Ig (600 mg/kg/month) and regular suppressive antimicrobials are the best means for controlling progression of lung disease, however, randomized clinical trials are needed for the exact proof Citation[68,98]. Reducing contact with fungal sources should be noted by patients, especially Aspergillus spp.-infected areas Citation[99].
Follow-up
Although there is no consensus for monitoring lung damage in CVID patients, pulmonary lung function tests (spirometery and carbon monoxide diffusion; at baseline and every 1–2 years) and high-resolution computed tomography (gold standard test; at baseline and every 4–5 years) are the best recommended examinations. Plain chest X-ray radiography is of limited value in CVID; however, it should be considered if the patient is febrile, has pleuritic pain and signs of consolidation, effusion or collapse. Moreover, because of the probability of radiosensitivity in some CVID cases, lower intervals with other X-ray procedures should be avoided as screening leads to excessive radiation exposure over time Citation[48,51,100–102]. Biopsy and pathological investigation should be carried out if large or persistent nodules are found in the lungs, which may change our therapeutic strategy to treat for polyclonal lymphocytic infiltrative disease-like granulomatous infiltrates Citation[53,103].
Treatment
Endoscopic sinus surgery may be required in cases of chronic sinusitis. Obstructive airway diseases of CVID patients can be managed with inhaled corticosteroids Citation[99]. There are several studies that report methods for preserving and improving lung function in pulmonary complicated CVID cases, including postural drainage, inspiratory muscle training and pulmonary rehabilitation programs Citation[104,105], advocating higher IgG trough levels (700–800 mg/dl) Citation[27], using anti-inflammatory effects of macrolides with azithromycin Citation[106–108], inhaled corticosteroids fluticasone or nebulized gentamicin for reduction of sputum production Citation[109,110], using an oral quinolone and aerosolized colimycin or tobramycin for aggressive eradication of colonized Pseudomonas spp. Citation[111,112], NSAIDs Citation[113] and mucolytics Citation[114]. Of interest, patients with bronchiectasis required IVIG higher than 600 mg/dl to achieve the same IgG level compared with patientswithout bronchiectasis Citation[115,116]. Clinical response, lung function and sputum sampling should be performed for respiratory health monitoring after starting the treatment.
In CVID patients with pulmonary diseases, new therapeutic approaches focus on IL-2 therapy Citation[117–120], short and long-acting inhaled B2-agonists in bronchiectasis Citation[121,122] and leukotriene receptor antagonists Citation[113]. Although most bronchiectatic CVID patients tend to have generalized lung damage, selected cases with highly localized disease may benefit from surgical procedure Citation[123].
Lung insufficiency due to progressive intractable pulmonary disease necessitates continuous oxygen treatment and/or heart or lung transplantation, although overall outcomes of this modality are variable in CVID cases Citation[53,124–126].
Management of polyclonal lymphocytic infiltrative complications
Approximately 10–25% of CVID patients encounter various lymphoproliferative and granulomatous diseases and the mean age at onset of these complications is 20–40 years. Granulomatous lesions can affect any organ in the progression of CVID, but the most common site is the lung, presenting with a pattern of interstitial lung disease (granulomatous lymphocytic interstitial lung disease [GLILD]) Citation[48,103,127,128].
The frequency of multisystemic involvement is unknown and probably underestimated Citation[129]. All CVID patients with pulmonary granulomatous diseases suffer from shortness of breath and persistent cough. Although physiological functions are normal in patients with pulmonary granulomas, it is not excluded that these patients would have eventually developed abnormal tests over time Citation[48]. Lymphoid interstitial pneumonitis (LIP) is another involvement of the pulmonary system in lymphoproliferative disorders without evidence to be related to any known pulmonary infection and it is associated with other connective tissue disorders Citation[36]. LIP and GLILD complications have a poor outcome and many unanswered questions about the pathogenesis and appropriate therapeutic intervention of both complications remain Citation[130].
The lymphoproliferative and granulomatous diseases in CVID seem to be related to severe depletion of switched memory B cells and CD4+ native T cells Citation[131]. Splenomegaly and malignancies are also more frequent in this subgroup of CVID patients Citation[48]. Expansion of CD8+ cells and lack of CD4+ cells has already been shown to be associated with the granuloma formation in tissues Citation[132–134]. Genetic predisposition may have a role in different aspects of granulomatous diseases in CVID patients.
Prevention
Ig replacement therapy has no effect on polyclonal lymphocytic infiltrative diseases as a preventive agent for inflammatory process that is made by an immune dysregulation mechanism Citation[40,135].
Follow-up
Biopsy via open lung or transbronchial surgery may be required for large and persistent lymph nodes. Signs of interstitial lung disease including dyspnea and reduced exercise tolerance should be considered in regular visits. Restrictive ventilatory pattern in pulmonary function test and parenchymal nodules or ground glass opacities in high-resolution computed tomography may alert physicians to consider pulmonary lymphoproliferative diseases. However, other imaging modalities (e.g., MRI, endoscopy) for assessment of progression of granulomatous diseases may be indicated.
Treatment
The optimum therapeutic option for CVID-related lymphoproliferative diseases is not clear, and this complication remains a major clinical challenge. High doses of systemic corticosteroids (10 mg a day or 20 mg every other day or twice-daily inhaled beclomethasone) is the first choice for interstitial lung disease with restrictive ventilatory defect due to lymphoproliferative disease such as pulmonary granulomas and LIP, but their long-term use is limited because of the risk of infections Citation[136,137]. For long-term therapy, hydroxychloroquine is prescribed with dosage of 200–400 mg a day (range: 3.5–6.5 mg/kg) Citation[138–140]. Steroid-sparing immunosuppressive agents have also been recommended in special situations when inflammation is predominantly pulmonary, including cyclosporine A (125 mg a day) Citation[135], methotrexate Citation[141], azathioprine, mycophenolate mofetil and 6-mercaptopurine Citation[136]. All mentioned therapeutic modalities are used in case reports or limited studies and their results for extrapolation to CVID should be tested in clinical trials. Monoclonal antibodies as TNF-α inhibitors, such as etanercept Citation[132,142] and infliximab Citation[143–146], can be used in the case of generalized granulomatous lesions combined with autoimmunity, but no systematic clinical trials have investigated their efficacy and safety. Opportunistic infections like Pneumocystis jirovecii pneumonia may result when patients are under treatment for lymphoproliferative diseases Citation[52,115].
Hepatomegaly and lymphadenopathy have also not been proven for treatment recommendations. Splenectomy should be avoided because its efficacy in the treatment of cytopenia is controversial and this surgery predisposes patients to severe infections Citation[4,147,148].
Management of autoimmune complications
Autoimmune-associated disorders, reported in 20–25% of CVID patients, are caused by immune dysregulation due to failed or circumvented specific autoreactivity checkpoints during the B-cell development process Citation[149]. This dysregulation culminates in production of multiple autoantibodies against various self-antigenic targets Citation[150]. Autoimmune thrombocytopenic purpura (ITP) and autoimmune hemolytic anemia (AIHA) are the most common types of autoimmune consequences, occurring in 5–8% of the all registered CVID patients Citation[4,151]. These disorders present in some patients before the diagnosis of CVID. Thus, CVID must be considered as a differential diagnosis in adult-onset ITP and AIHA Citation[152,153]. Other autoimmune disorders include the presence of anti-IgA antibodies Citation[154], autoimmune neutropenia Citation[36], pernicious anemia Citation[140,150], juvenile rheumatoid arthritis, systemic lupus erythematosus, psoriasis, autoimmune thyroiditis, autoimmune hepatitis, primary biliary cirrhosis, insulin dependent diabetes, sicca syndrome, vitiligo and vasculitis Citation[25,36,155,156].
Prevention
No preventive modalities are presently available, however, IVIG treatment has led to reduction of incidence of the autoimmune diseases especially within cases with ITP and AIHA Citation[153].
Follow-up
General medical vigilance and hematologic laboratory monitoring with intervals of 3–6 months is the way for monitoring of hematologic autoimmunity. Some clinical immunologists recommend annual thyroid examination and thyroid function testing as part of routine follow-up in CVID patients.
Treatment
Common treatment protocols for autoimmunity are used similarly in CVID cases. In case of hematologic autoimmunity, managements are based on intravenous corticosteroids (1 g of methylprednisolone in mild disease) or anti CD-20 monoclonal antibodies (rituximab in severe disorders) Citation[36,157,158]. Persistent autoimmunity can be handled by high Ig immunomodulatory effects (1 g/kg/week) for a short time.
TNF-α inhibitors might be used in overlapped phenotypes of autoimmunity (e.g., infliximab for Crohn’s disease and etanercept for rheumatoid arthritis) and polyclonal lymphocytic infiltration Citation[142,159].
Management of GI complications & enteropathy
The GI tract constitutes the largest human immune organ and is therefore expected to be affected by CVID Citation[55,160–162]. Virtually 60% of CVID patients present with diarrhea and 10% develop digestive complications such as idiopathic malabsorption associated with weight loss Citation[163]. Other gut symptoms such as discomfort and bloating are also common Citation[25]. Inflammatory GI disorders are extremely frequent in CVID patients Citation[164]. Approximately 20% of CVID patients have GI symptoms not related to infectious causes Citation[4]. Classical Crohn’s disease is probably not seen in CVID, although it has a high incidence in X-linked agamamglobulinemia. Celiac (gluten-sensitive) disease does occur but is very rare. CVID-related enteropathy is a sprue-like illness with villus atrophy and crypt hyperplasia in biopsy but shows no improvement by gluten-free diet Citation[165]. Inflammatory bowel disease (ulcerative colitis, ulcerative proctitis or Crohn’s disease), which can cause stricture, lymphocytic colitis and collagen enterocolitis, is more prevalent in CVID patients than the normal population Citation[166]. Intestinal lymphangiectasia, nodular lymphoid hyperplasia and nonspecific malabsorption have also been reported Citation[4]. Defective cellular immunity, rather than antibody deficiency alone, appears to predispose patients to such symptoms Citation[164]. Up to 10% of CVID patients may suffer from liver dysfunctions (abnormal liver function tests, predominantly increased alkaline phosphatase and nodular regenerative hyperplasia) caused by infections (e.g., HBV and HCV), autoimmunity (e.g., autoimmune hepatitis, primary biliary cirrhosis) and granulomatous diseases Citation[84,160,167]. Previous studies suggested that nodular regenerative hyperplasia has different etiology, including circulating immune complexes or light chain deposits in the sinusoidal walls Citation[168]. Unexplained liver disease with portal hypertension occurs in 5–10% of the patients. Autoimmunity (69.2%) and lymphocyte abnormalities (78.6%) were observed more frequently in CVID patients with hepatic dysfunctions Citation[169–173].
Prevention
No effective preventive modalities are presently available for GI complications. However, prevention of primary hepatic complications (viral hepatitis, autoimmunity and granulomatous diseases) helps progression of CVID cases from regenerative hyperplasia leading to portal hypertension, cholestasis and hepatic dysfunction. It remains to be proven whether fast treatment of intestinal infections improves outcomes for CVID patients Citation[14].
Follow-up
Screening for GI complications is more unclear, unless attention is paid to the patient complaints (diarrhea refractory to antibiotic treatments) and clinical symptoms (especially weight loss) that are key alarm signs Citation[14]. Biannual upper and/or lower endoscopy and yearly ultrasonograohy may help screening for GI symptomatic CVID cases.
Treatment
The management of severe inflammatory enteropathy in CVID is based on low-dose immunomodulators (azathioprine or 6-mercaptopurine) and TNF-α blockers (infliximab or etanercept) Citation[165,174]. Management in mild inflammatory bowel disease, however, is the same as for immunocompetent patients. The use of long-term high-dose corticosteroids is controversial because of the increased probability of intestinal CMV infection Citation[160,175].
Management of malignancy
CVID patients are at higher risk of neoplasias (hematological or solid tumors) compared with normal population (over ten-times the risk) Citation[176]. The most common type of malignancy is non-Hodgkin’s lymphoma (NHL), which is more likely to be of B-cell origin Citation[177]. Lymphoid malignancies (including mucosa-associated lymphoid tissue lymphoma, marginal zone lymphoma and T-cell-rich B-cell EBV-associated lymphoma) are more prevalent in younger patients Citation[5,34,178]; by contrast, CVID adults are more susceptible to GI tract malignancies, especially adenocarcinoma of the stomach Citation[177]. It was estimated that 8.2% of CVID cases are susceptible for lymphoma, however, females and CVID cases with higher levels of serum IgM are more prone to this secondary complication Citation[26]. Polyclonal lymphocytic infiltration is a clinical predictor associated with an increased risk of lymphoid malignancy Citation[51]. Furthermore, in CVID patients, clinical and family histories of neoplasia should be taken accurately along with consideration of surveillance for malignancy, especially lymphoma and gastric cancer Citation[48]. Risk of gastric cancer, especially gastric adenocarcinoma, may be increased by H. pylori infections and chromosomal radiosensitivity Citation[177,179]. Chronic viral infections with CMV and human herpesvirus 8 may predispose CVID patients to develop NHL Citation[176].
Prevention
H. pylori antigen screening in feces and appropriate endoscopy with examination for this microorganism and its eradication may prevent gastric cancer Citation[180]. Moreover, decreasing unnecessary exposure of CVID patients to irradiation can reduce risk of iatrogenic cancer due to chromosomal radiosensitivity Citation[177].
Follow-up
First of all, age-appropriate cancer screening of general healthy population (colonoscopy, prostate examination, cervical smears and mammograms) should be programmed for all CVID patients more tenuously. CVID-specific screening by endoscopy for finding mucosal changes should be performed in symptomatic cases. Histopathological investigation of enlarged nodes via excision of the whole lymph node and periodic complete blood counts and differential white blood cell counts are important means for screening of lymphoid malignancy Citation[181]. Bone marrow examinations for lymphoma screening, however, are not positive, except in the most advanced cases.
Treatment
Management of neoplasia in CVID is similar to routine chemotherapy protocols for cancerous patients as well as standard rituximab protocols. Surgical modalities such as total gastrectomy are lifesaving for early diagnosed cancers Citation[177].
Because of the relation of presentation of malignancy and impaired T-cell immunity in CVID, allogeneic stem cell transplantation is now considered in these selected CVID cases. However, it should be noticed that these potentially curative approaches are experimental and should only be proposed for therapy-refractory life-threatening complications (late-onset combined immunodeficiency subgroup) with careful process for donor selection Citation[182]. The best response of allogeneic stem cell transplantation is seen in the patients with NHL resolving all CVID-related consequences. Graft-versus-host disease should be monitored in these patients Citation[183].
Prognosis & surveillance
Major indicators that affect poor CVID prognosis are structural pulmonary damages, GLILD, severe autoimmunity, malignancy and extent of end-organ damage, which all can be managed by therapeutic strategies Citation[103]. Moreover, implementation of appropriate prevention, screening and treatment protocols during recent years improved mortality rate after 10 years of follow-up from 20–40% Citation[4,33] to 5–10% Citation[13,34]. Respiratory tract insufficiency, especially cor pulmonale, serves as the most frequent cause of death in CVID patients followed by lymphoma and liver failure. Despite clinical manifestations, low levels of IgG, poor T-cell responses to antigens, and a low percentage of peripheral B cells are laboratory factors that can predict lower survival rates in CVID.
Expert commentary
Ig replacement therapy, either subcutaneous or intravenous, is the mainstay of therapy in patients with CVID, while specific treatment for certain complications associated with disease is needed. In addition to recurrent infections, in which antibiotic prophylaxis might be added to Ig replacement therapy, further pulmonary problems as well as polyclonal lymphocytic infiltrative complications, enteropathy, autoimmune diseases and malignancies require additional management, and higher doses of Ig replacement therapy can be recommended in certain conditions.
Five-year view
It is to be hoped that underlying genetic defect(s) and precise pathophysiology of CVID will be identified in the near future, considering multicenter international studies in this field, which could provide novel possibilities for treatment of this disease. Moreover, conducting some studies on safety and efficacy of current treatments can help clinicians come to a consensus on treatment of this disease, which definitely can improve the prognosis of the patients.
Table 1. Abstracted guideline for management of common variable immunodeficiency complications.
Key issues
• Education of patients, immunoglobulin replacement therapy, prophylactic and therapeutic antibiotics and complementary vaccinations are the main means for tackling infectious complications of common variable immunodeficiency (CVID) cases.
• Pulmonary function tests and high-resolution computed tomography should be considered in all CVID patients to diagnose secondary lung damages at the earliest time possible.
• Annual ultrasonography and endoscopy every 2 years are the best screening methods for individuals with suspected gut complication.
• Steroid-sparing immunosuppressive agents and TNF-α inhibitors may be useful therapeutic modalities in patients with lymphoproliferative and autoimmune phenotypes.
• Beside routine protocols of chemotherapy, allogeneic stem cell transplantation may have an appropriate result in malignant CVID patients.
References
- Notarangelo LD, Fischer A, Geha RS et al. Primary immunodeficiencies: 2009 update. J. Allergy Clin. Immunol. 124(6), 1161–1178 (2009).
- Notarangelo LD. Primary immunodeficiencies. J. Allergy Clin. Immunol. 125(2 Suppl. 2), S182–S194 (2010).
- Aghamohammadi A, Farhoudi A, Moin M et al. Clinical and immunological features of 65 Iranian patients with common variable immunodeficiency. Clin. Diagn. Lab. Immunol. 12(7), 825–832 (2005).
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin. Immunol. 92(1), 34–48 (1999).
- Cunningham-Rundles C, Siegal FP, Cunningham-Rundles S, Lieberman P. Incidence of cancer in 98 patients with common varied immunodeficiency. J. Clin. Immunol. 7(4), 294–299 (1987).
- Ko J, Radigan L, Cunningham-Rundles C. Immune competence and switched memory B cells in common variable immunodeficiency. Clin. Immunol. 116(1), 37–41 (2005).
- Darragh PM. The prevalence and prevention of severe mental handicap in Northern Ireland. Ir. Med. J. 75(1), 16–19 (1982).
- Fasth A. Primary immunodeficiency disorders in Sweden: cases among children, 1974-1979. J. Clin. Immunol. 2(2), 86–92 (1982).
- Hammarström L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID). Clin. Exp. Immunol. 120(2), 225–231 (2000).
- Luzi G, Businco L, Aiuti F. Primary immunodeficiency syndromes in Italy: a report of the national register in children and adults. J. Clin. Immunol. 3(4), 316–320 (1983).
- Chapel H, Geha R, Rosen F; IUIS PID (Primary Immunodeficiencies) Classification committee. Primary immunodeficiency diseases: an update. Clin. Exp. Immunol. 132(1), 9–15 (2003).
- Notarangelo L, Casanova JL, Fischer A et al.; International Union of Immunological Societies Primary Immunodeficiency diseases classification committee. Primary immunodeficiency diseases: an update. J. Allergy Clin. Immunol. 114(3), 677–687 (2004).
- Chapel H, Lucas M, Lee M et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 112(2), 277–286 (2008).
- Cunningham-Rundles C. How I treat common variable immune deficiency. Blood 116(1), 7–15 (2010).
- Janeway CA, Apt L, Gitlin D. Agammaglobulinemia. Trans. Assoc. Am. Physicians 66, 200–202 (1953).
- Grimbacher B, Warnatz K, Peter HH. The immunological synapse for B-cell memory: the role of the ICOS and its ligand for the longevity of humoral immunity. Curr. Opin. Allergy Clin. Immunol. 3(6), 409–419 (2003).
- Grimbacher B, Hutloff A, Schlesier M et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat. Immunol. 4(3), 261–268 (2003).
- van Zelm MC, Reisli I, van der Burg M et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N. Engl. J. Med. 354(18), 1901–1912 (2006).
- Lopez-Herrera G, Tampella G, Pan-Hammarström Q et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am. J. Hum. Genet. 90(6), 986–1001 (2012).
- Kuijpers TW, Bende RJ, Baars PA et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J. Clin. Invest. 120(1), 214–222 (2010).
- van Zelm MC, Smet J, Adams B et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J. Clin. Invest. 120(4), 1265–1274 (2010).
- Frank MM. CD21 deficiency, complement, and the development of common variable immunodeficiency. J. Allergy Clin. Immunol. 129(3), 811–813 (2012).
- Thiel J, Kimmig L, Salzer U et al. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J. Allergy Clin. Immunol. 129(3), 801–810.e6 (2012).
- Salzer U, Warnatz K, Peter HH. Common variable immunodeficiency – an update. Arthritis Res. Ther. 14(5), 223 (2012).
- Yong PF, Tarzi M, Chua I, Grimbacher B, Chee R. Common variable immunodeficiency: an update on etiology and management. Immunol. Allergy Clin. North Am. 28(2), 367–386, ix (2008).
- Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 119(7), 1650–1657 (2012).
- Fried AJ, Bonilla FA. Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections. Clin. Microbiol. Rev. 22(3), 396–414 (2009).
- Aghamohammadi A, Parvaneh N, Rezaei N. Common variable immunodeficiency: a heterogeneous group needs further subclassification. Expert Rev. Clin. Immunol. 5(6), 629–631 (2009).
- Aghamohammadi A, Kanegane H, Moein M et al. Identification of an SH2D1A mutation in a hypogammaglobulinemic male patient with a diagnosis of common variable immunodeficiency. Int. J. Hematol. 78(1), 45–47 (2003).
- Oraei M, Aghamohammadi A, Rezaei N et al. Naive CD4+ T cells and recent thymic emigrants in common variable immunodeficiency. J. Investig. Allergol. Clin. Immunol. 22(3), 160–167 (2012).
- Rezaei N, Aghamohammadi A, Nourizadeh M et al. Cytokine production by activated T cells in common variable immunodeficiency. J. Investig. Allergol. Clin. Immunol. 20(3), 244–251 (2010).
- Salzer U, Maul-Pavicic A, Cunningham-Rundles C et al. ICOS deficiency in patients with common variable immunodeficiency. Clin. Immunol. 113(3), 234–240 (2004).
- Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Q. J. Med. 86(1), 31–42 (1993).
- Quinti I, Soresina A, Spadaro G et al.; Italian Primary Immunodeficiency Network. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J. Clin. Immunol. 27(3), 308–316 (2007).
- Bonilla FA, Bernstein IL, Khan DA et al.; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann. Allergy Asthma Immunol. 94(5 Suppl. 1), S1–S63 (2005).
- Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br. J. Haematol. 145(6), 709–727 (2009).
- Abolhassani H, Aghamohammadi A, Abolhassani F, Eftekhar H, Heidarnia M, Rezaei N. Health policy for common variable immunodeficiency: burden of the disease. J. Investig. Allergol. Clin. Immunol. 21(6), 454–458 (2011).
- Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 109(6), 1001–1004 (2002).
- Björkander J, Wadsworth C, Hanson LA. 1040 prophylactic infusions with an unmodified intravenous immunoglobulin product causing few side-effects in patients with antibody deficiency syndromes. Infection 13(3), 102–110 (1985).
- de Gracia J, Vendrell M, Alvarez A et al. Immunoglobulin therapy to control lung damage in patients with common variable immunodeficiency. Int. Immunopharmacol. 4(6), 745–753 (2004).
- Pourpak Z, Aghamohammadi A, Sedighipour L et al. Effect of regular intravenous immunoglobulin therapy on prevention of pneumonia in patients with common variable immunodeficiency. J. Microbiol. Immunol. Infect. 39(2), 114–120 (2006).
- Warnatz K, Denz A, Dräger R et al. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood 99(5), 1544–1551 (2002).
- Wehr C, Kivioja T, Schmitt C et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood 111(1), 77–85 (2008).
- Piqueras B, Lavenu-Bombled C, Galicier L et al. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J. Clin. Immunol. 23(5), 385–400 (2003).
- Giovannetti A, Pierdominici M, Mazzetta F et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J. Immunol. 178(6), 3932–3943 (2007).
- Malphettes M, Gérard L, Carmagnat M et al.; DEFI Study Group. Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin. Infect. Dis. 49(9), 1329–1338 (2009).
- Aghamohammadi A, Abolhassani H, Moazzami K, Parvaneh N, Rezaei N. Correlation between common variable immunodeficiency clinical phenotypes and parental consanguinity in children and adults. J. Investig. Allergol. Clin. Immunol. 20(5), 372–379 (2010).
- Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet 372(9637), 489–502 (2008).
- Oksenhendler E, Gérard L, Fieschi C et al.; DEFI Study Group. Infections in 252 patients with common variable immunodeficiency. Clin. Infect. Dis. 46(10), 1547–1554 (2008).
- Aghamohammadi A, Moazzami K, Rezaei N et al. ENT manifestations in Iranian patients with primary antibody deficiencies. J. Laryngol. Otol. 122(4), 409–413 (2008).
- Hampson FA, Chandra A, Screaton NJ et al. Respiratory disease in common variable immunodeficiency and other primary immunodeficiency disorders. Clin. Radiol. 67(6), 587–595 (2012).
- Martínez García MA, de Rojas MD, Nauffal Manzur MD et al. Respiratory disorders in common variable immunodeficiency. Respir. Med. 95(3), 191–195 (2001).
- Cunningham-Rundles C. Common variable immunodeficiency. Curr. Allergy Asthma Rep. 1(5), 421–429 (2001).
- Onbasi K, Günsar F, Sin AZ, Ardeniz O, Kokuludag A, Sebik F. Common variable immunodeficiency (CVID) presenting with malabsorption due to giardiasis. Turk. J. Gastroenterol. 16(2), 111–113 (2005).
- Kalha I, Sellin JH. Common variable immunodeficiency and the gastrointestinal tract. Curr. Gastroenterol. Rep. 6(5), 377–383 (2004).
- Zullo A, Romiti A, Rinaldi V et al. Gastric pathology in patients with common variable immunodeficiency. Gut 45(1), 77–81 (1999).
- Aghamohammadi A, Abolhassani H, Hirbod-Mobarakeh A et al. The uncommon combination of common variable immunodeficiency, macrophage activation syndrome, and cytomegalovirus retinitis. Viral Immunol. 25(2), 161–165 (2012).
- Rezaei N, Abolhassani H, Aghamohammadi A, Ochs HD. Indications and safety of intravenous and subcutaneous immunoglobulin therapy. Expert Rev. Clin. Immunol. 7(3), 301–316 (2011).
- Marashi SM, Raeiszadeh M, Enright V et al. Influence of cytomegalovirus infection on immune cell phenotypes in patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 129(5), 1349–1356.e3 (2012).
- Rotbart HA, Webster AD; Pleconaril Treatment Registry Group. Treatment of potentially life-threatening enterovirus infections with pleconaril. Clin. Infect. Dis. 32(2), 228–235 (2001).
- Halliday E, Winkelstein J, Webster AD. Enteroviral infections in primary immunodeficiency (PID): a survey of morbidity and mortality. J. Infect. 46(1), 1–8 (2003).
- van de Ven AA, Douma JW, Rademaker C et al. Pleconaril-resistant chronic parechovirus-associated enteropathy in agammaglobulinaemia. Antivir. Ther. 16(4), 611–614 (2011).
- Weiler CR. Immunoglobulin therapy: history, indications, and routes of administration. Int. J. Dermatol. 43(3), 163–166 (2004).
- Salehzadeh M, Aghamohammadi A, Rezaei N. Evaluation of immunoglobulin levels and infection rate in patients with common variable immunodeficiency after immunoglobulin replacement therapy. J. Microbiol. Immunol. Infect. 43(1), 11–17 (2010).
- Koleba T, Ensom MH. Pharmacokinetics of intravenous immunoglobulin: a systematic review. Pharmacotherapy 26(6), 813–827 (2006).
- Yong PL, Boyle J, Ballow M et al. Use of intravenous immunoglobulin and adjunctive therapies in the treatment of primary immunodeficiencies: A working group report of and study by the Primary Immunodeficiency Committee of the American Academy of Allergy Asthma and Immunology. Clin. Immunol. 135(2), 255–263 (2010).
- Orange JS, Hossny EM, Weiler CR et al.; Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. Use of intravenous immunoglobulin in human disease: a review of evidence by members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. J. Allergy Clin. Immunol. 117(Suppl. 4), S525–S553 (2006).
- Eijkhout HW, van Der Meer JW, Kallenberg CG et al.; Inter-University Working Party for the Study of Immune Deficiencies. The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia. A randomized, double-blind, multicenter crossover trial. Ann. Intern. Med. 135(3), 165–174 (2001).
- Moore ML, Quinn JM. Subcutaneous immunoglobulin replacement therapy for primary antibody deficiency: advancements into the 21st century. Ann. Allergy Asthma Immunol. 101(2), 114–121; quiz 122 (2008).
- Chapel HM, Spickett GP, Ericson D, Engl W, Eibl MM, Bjorkander J. The comparison of the efficacy and safety of intravenous versus subcutaneous immunoglobulin replacement therapy. J. Clin. Immunol. 20(2), 94–100 (2000).
- Abolhassani H, Sadaghiani MS, Aghamohammadi A, Ochs HD, Rezaei N. Home-based subcutaneous immunoglobulin versus hospital-based intravenous immunoglobulin in treatment of primary antibody deficiencies: systematic review and meta analysis. J. Clin. Immunol. 32(6), 1180–1192 (2012).
- Berger M; Flebogamma 5% DIF Investigators. A multicenter, prospective, open label, historically controlled clinical trial to evaluate efficacy and safety in primary immunodeficiency diseases (PID) patients of Flebogamma 5% DIF, the next generation of Flebogamma. J. Clin. Immunol. 27(6), 628–633 (2007).
- Dashti-Khavidaki S, Aghamohammadi A, Farshadi F et al. Adverse reactions of prophylactic intravenous immunoglobulin; a 13-year experience with 3004 infusions in Iranian patients with primary immunodeficiency diseases. J. Investig. Allergol. Clin. Immunol. 19(2), 139–145 (2009).
- Aghamohammadi A, Farhoudi A, Nikzad M et al. Adverse reactions of prophylactic intravenous immunoglobulin infusions in Iranian patients with primary immunodeficiency. Ann. Allergy Asthma Immunol. 92(1), 60–64 (2004).
- Khan S, Abuzakouk M, Doré PC, Sewell WA. Administering intravenous immunoglobulin during infection is associated with infusion reactions in selected patients. Ir. J. Med. Sci. 180(1), 125–128 (2011).
- Maarschalk-Ellerbroek LJ, Hoepelman IM, Ellerbroek PM. Immunoglobulin treatment in primary antibody deficiency. Int. J. Antimicrob. Agents 37(5), 396–404 (2011).
- Brinker KA, Silk HJ. Common variable immune deficiency and treatment with intravenous immunoglobulin during pregnancy. Ann. Allergy Asthma Immunol. 108(6), 464–465 (2012).
- Stein MR, Nelson RP, Church JA et al.; IgPro10 in PID study group. Safety and efficacy of Privigen, a novel 10% liquid immunoglobulin preparation for intravenous use, in patients with primary immunodeficiencies. J. Clin. Immunol. 29(1), 137–144 (2009).
- Brennan VM, Salomé-Bentley NJ, Chapel HM; Immunology Nurses Study. Prospective audit of adverse reactions occurring in 459 primary antibody-deficient patients receiving intravenous immunoglobulin. Clin. Exp. Immunol. 133(2), 247–251 (2003).
- de Albuquerque Campos R, Sato MN, da Silva Duarte AJ. IgG anti-IgA subclasses in common variable immunodeficiency and association with severe adverse reactions to intravenous immunoglobulin therapy. J. Clin. Immunol. 20(1), 77–82 (2000).
- Burks AW, Sampson HA, Buckley RH. Anaphylactic reactions after gamma globulin administration in patients with hypogammaglobulinemia. Detection of IgE antibodies to IgA. N. Engl. J. Med. 314(9), 560–564 (1986).
- Sati HI, Ahya R, Watson HG. Incidence and associations of acute renal failure complicating high-dose intravenous immunoglobulin therapy. Br. J. Haematol. 113(2), 556–557 (2001).
- Saroukhani S, Aghamohammadi A, Mahmoudi-Gharaei J et al. Behavior abnormality following intravenous immunoglobulin treatment in patients with primary antibody deficiencies. Hum. Psychopharmacol. 25(5), 419–422 (2010).
- Lindberg K, Gustafson R, Samuelson A, Rynnel-Dagöö B. Impact of IgG replacement therapy and antibiotic treatment on the colonization of non-encapsulated Haemophilus influenzae in the nasopharynx in patients with hypogammaglobulinaemia. Scand. J. Infect. Dis. 33(12), 904–908 (2001).
- Stirling RG. Primary immunodeficiency: a call for multidisciplinary care. Lancet 372(9653), 1877–1878; author reply 1878 (2008).
- Sewell WA, Buckland M, Jolles SR. Therapeutic strategies in common variable immunodeficiency. Drugs 63(13), 1359–1371 (2003).
- López-Boado YS, Rubin BK. Macrolides as immunomodulatory medications for the therapy of chronic lung diseases. Curr. Opin. Pharmacol. 8(3), 286–291 (2008).
- Luisi F, Gandolfi TD, Daudt AD, Sanvitto JP, Pitrez PM, Pinto LA. Anti-inflammatory effects of macrolides in childhood lung diseases. J. Bras. Pneumol. 38(6), 786–796 (2012).
- Langelot M, Cellerin L, Germaud P. Anti-inflammatory effects of macrolides: applications in lung disease. Rev. Pneumol. Clin. 62(4), 215–222 (2006).
- Resnick E, Cunningham-Rundles C. Treatment of common variable immune deficiency. Exp. Opin. Orphan Drugs 1(2), 157–166 (2013).
- Kroger AT, Atkinson WL, Marcuse EK, Pickering LK; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR. Recomm. Rep. 55(RR-15), 1–48 (2006).
- Atkinson WL, Pickering LK, Schwartz B, Weniger BG, Iskander JK, Watson JC; Centers for Disease Control and Prevention. General recommendations on immunization. Recommendations of the Advisory Committee on Immunization Practices (ACIP) and the American Academy of Family Physicians (AAFP). MMWR. Recomm. Rep. 51(RR-2), 1–35 (2002).
- Serana F, Airò P, Chiarini M et al. Thymic and bone marrow output in patients with common variable immunodeficiency. J. Clin. Immunol. 31(4), 540–549 (2011).
- Nakagawa N, Imai K, Kanegane H et al. Quantification of k-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J. Allergy Clin. Immunol. 128(1), 223–225.e2 (2011).
- Medlicott SA, Coderre S, Horne G, Panaccione R. Multimodal immunosuppressant therapy in steroid-refractory common variable immunodeficiency sprue: a case report complicating cytomegalovirus infection. Int. J. Surg. Pathol. 14(1), 101–106 (2006).
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA 279(1), 58–61 (1998).
- Tanaka N, Kim JS, Bates CA et al. Lung diseases in patients with common variable immunodeficiency: chest radiographic, and computed tomographic findings. J. Comput. Assist. Tomogr. 30(5), 828–838 (2006).
- Roifman CM, Levison H, Gelfand EW. High-dose versus low-dose intravenous immunoglobulin in hypogammaglobulinaemia and chronic lung disease. Lancet 1(8541), 1075–1077 (1987).
- Quartier P, Debré M, De Blic J et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: a retrospective survey of 31 patients. J. Pediatr. 134(5), 589–596 (1999).
- Aghamohammadi A, Moin M, Kouhi A et al. Chromosomal radiosensitivity in patients with common variable immunodeficiency. Immunobiology 213(5), 447–454 (2008).
- Palanduz S, Palanduz A, Yalcin I et al. In vitro chromosomal radiosensitivity in common variable immune deficiency. Clin. Immunol. Immunopathol. 86(2), 180–182 (1998).
- Vorechovský I, Scott D, Haeney MR, Webster DA. Chromosomal radiosensitivity in common variable immune deficiency. Mutat. Res. 290(2), 255–264 (1993).
- Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J. Allergy Clin. Immunol. 114(2), 415–421 (2004).
- Garrod R, Lasserson T. Role of physiotherapy in the management of chronic lung diseases: an overview of systematic reviews. Respir. Med. 101(12), 2429–2436 (2007).
- Bradley J, Moran F, Greenstone M. Physical training for bronchiectasis. Cochrane Database Syst. Rev. 3, CD002166 (2002).
- Yalçin E, Kiper N, Ozçelik U et al. Effects of claritromycin on inflammatory parameters and clinical conditions in children with bronchiectasis. J. Clin. Pharm. Ther. 31(1), 49–55 (2006).
- Koh YY, Lee MH, Sun YH, Sung KW, Chae JH. Effect of roxithromycin on airway responsiveness in children with bronchiectasis: a double-blind, placebo-controlled study. Eur. Respir. J. 10(5), 994–999 (1997).
- Cymbala AA, Edmonds LC, Bauer MA et al. The disease-modifying effects of twice-weekly oral azithromycin in patients with bronchiectasis. Treat. Respir. Med. 4(2), 117–122 (2005).
- Lin HC, Cheng HF, Wang CH, Liu CY, Yu CT, Kuo HP. Inhaled gentamicin reduces airway neutrophil activity and mucus secretion in bronchiectasis. Am. J. Respir. Crit. Care Med. 155(6), 2024–2029 (1997).
- Tsang KW, Tan KC, Ho PL et al. Inhaled fluticasone in bronchiectasis: a 12 month study. Thorax 60(3), 239–243 (2005).
- Jensen T, Pedersen SS, Garne S, Heilmann C, Høiby N, Koch C. Colistin inhalation therapy in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. J. Antimicrob. Chemother. 19(6), 831–838 (1987).
- Chuchalin A, Csiszér E, Gyurkovics K et al. A formulation of aerosolized tobramycin (Bramitob) in the treatment of patients with cystic fibrosis and Pseudomonas aeruginosa infection: a double-blind, placebo-controlled, multicenter study. Paediatr. Drugs 9(Suppl. 1), 21–31 (2007).
- Corless JA, Warburton CJ. Surgery vs non-surgical treatment for bronchiectasis. Cochrane Database Syst. Rev. 4, CD002180 (2000).
- O’Donnell AE, Barker AF, Ilowite JS, Fick RB. Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNase I. rhDNase Study Group. Chest 113(5), 1329–1334 (1998).
- Touw CM, van de Ven AA, de Jong PA et al. Detection of pulmonary complications in common variable immunodeficiency. Pediatr. Allergy Immunol. 21(5), 793–805 (2010).
- Busse PJ, Farzan S, Cunningham-Rundles C. Pulmonary complications of common variable immunodeficiency. Ann. Allergy Asthma Immunol. 98(1), 1–8; quiz 8 (2007).
- Foruny JR, Bárcena R, Moreno A et al. Benefit of pegylated interferon-alpha-2a/ribavirin in a patient with common variable immunodeficiency and hepatitis C virus cirrhosis after liver transplantation and splenic embolization. Transplantation 82(2), 289–290 (2006).
- Cunningham-Rundles C, Bodian C, Ochs HD, Martin S, Reiter-Wong M, Zhuo Z. Long-term low-dose IL-2 enhances immune function in common variable immunodeficiency. Clin. Immunol. 100(2), 181–190 (2001).
- Rump JA, Jahreis A, Schlesier M, Stecher S, Peter HH. A double-blind, placebo-controlled, crossover therapy study with natural human IL-2 (nhuIL-2) in combination with regular intravenous gammaglobulin (IVIG) infusions in 10 patients with common variable immunodeficiency (CVID). Clin. Exp. Immunol. 110(2), 167–173 (1997).
- Ten RM, Anderson PM, Zein NN, Temesgen Z, Clawson ML, Weiss W. Interleukin-2 liposomes for primary immune deficiency using the aerosol route. Int. Immunopharmacol. 2(2–3), 333–344 (2002).
- Franco F, Sheikh A, Greenstone M. Short acting beta-2 agonists for bronchiectasis. Cochrane Database Syst. Rev. 3, CD003572 (2003).
- Sheikh A, Nolan D, Greenstone M. Long-acting beta-2-agonists for bronchiectasis. Cochrane Database Syst. Rev. 4, CD002155 (2001).
- Corless JA, Warburton CJ. Surgery vs non-surgical treatment for bronchiectasis. Cochrane Database Syst. Rev. 4, CD002180 (2000).
- Bethune CA, Spickett GP. Common variable immunodeficiency: an update on therapeutic approaches. BioDrugs 13(4), 243–253 (2000).
- Burton CM, Milman N, Andersen CB, Marquart H, Iversen M. Common variable immune deficiency and lung transplantation. Scand. J. Infect. Dis. 39(4), 362–367 (2007).
- Hill AT, Thompson RA, Wallwork J, Stableforth DE. Heart lung transplantation in a patient with end stage lung disease due to common variable immunodeficiency. Thorax 53(7), 622–623 (1998).
- Morimoto Y, Routes JM. Granulomatous disease in common variable immunodeficiency. Curr. Allergy Asthma Rep. 5(5), 370–375 (2005).
- Chase NM, Verbsky JW, Hintermeyer MK et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J. Clin. Immunol. 33(1), 30–39 (2013).
- Aghamohammadi A, Abolhassani H, Rezaei N et al. Cutaneous granulomas in common variable immunodeficiency: case report and review of literature. Acta Dermatovenerol. Croat. 18(2), 107–113 (2010).
- Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin. Immunol. 134(2), 97–103 (2010).
- Bouvry D, Mouthon L, Brillet PY et al.; Groupe Sarcoïdose Francophone. Granulomatosis-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur. Respir. J. 41(1), 115–122 (2013).
- Lin JH, Liebhaber M, Roberts RL, Dyer Z, Stiehm ER. Etanercept treatment of cutaneous granulomas in common variable immunodeficiency. J. Allergy Clin. Immunol. 117(4), 878–882 (2006).
- Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin. Immunol. 133(2), 198–207 (2009).
- North ME, Webster AD, Farrant J. Primary defect in CD8+ lymphocytes in the antibody deficiency disease (common variable immunodeficiency): abnormalities in intracellular production of interferon-gamma (IFN-gamma) in CD28+ (‘cytotoxic’) and CD28- (‘suppressor’) CD8+ subsets. Clin. Exp. Immunol. 111(1), 70–75 (1998).
- Davies CW, Juniper MC, Gray W, Gleeson FV, Chapel HM, Davies RJ. Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinaemia treated with cyclosporin A. Thorax 55(1), 88–90 (2000).
- Paramothayan S, Lasserson T. Treatments for pulmonary sarcoidosis. Respir. Med. 102(1), 1–9 (2008).
- Boursiquot JN, Gérard L, Malphettes M et al.; DEFI study group. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J. Clin. Immunol. 33(1), 84–95 (2013).
- Fazzi P. Pharmacotherapeutic management of pulmonary sarcoidosis. Am. J. Respir. Med. 2(4), 311–320 (2003).
- Kyburz D, Brentano F, Gay S. Mode of action of hydroxychloroquine in RA – evidence of an inhibitory effect on toll-like receptor signaling. Nat. Clin. Pract. Rheumatol. 2(9), 458–459 (2006).
- Knight AK, Cunningham-Rundles C. Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmun. Rev. 5(2), 156–159 (2006).
- Paramothayan S, Lasserson TJ, Walters EH. Immunosuppressive and cytotoxic therapy for pulmonary sarcoidosis. Cochrane Database Syst. Rev. 3, CD003536 (2006).
- Smith KJ, Skelton H. Common variable immunodeficiency treated with a recombinant human IgG, tumour necrosis factor-alpha receptor fusion protein. Br. J. Dermatol. 144(3), 597–600 (2001).
- Fernández-Ruiz M, Guerra-Vales JM, Francisco-Javier CF et al. Fever of unknown origin in a patient with common variable immunodeficiency associated with multisystemic granulomatous disease. Intern. Med. 46(15), 1197–1202 (2007).
- Hatab AZ, Ballas ZK. Caseating granulomatous disease in common variable immunodeficiency treated with infliximab. J. Allergy Clin. Immunol. 116(5), 1161–1162 (2005).
- Baughman RP, Drent M, Kavuru M et al.; Sarcoidosis Investigators. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am. J. Respir. Crit. Care Med. 174(7), 795–802 (2006).
- Thatayatikom A, Thatayatikom S, White AJ. Infliximab treatment for severe granulomatous disease in common variable immunodeficiency: a case report and review of the literature. Ann. Allergy Asthma Immunol. 95(3), 293–300 (2005).
- Sève P, Bourdillon L, Sarrot-Reynauld F et al.; DEF-I Study Group. Autoimmune hemolytic anemia and common variable immunodeficiency: a case-control study of 18 patients. Medicine (Baltimore). 87(3), 177–184 (2008).
- Wong MS, Chan GC, Ha SY, Lau YL. Clinical characteristics of chronic idiopathic thrombocytopenia in Chinese children. J. Pediatr. Hematol. Oncol. 24(8), 648–652 (2002).
- Tsuiji M, Yurasov S, Velinzon K, Thomas S, Nussenzweig MC, Wardemann H. A checkpoint for autoreactivity in human IgM+ memory B cell development. J. Exp. Med. 203(2), 393–400 (2006).
- Brandt D, Gershwin ME. Common variable immune deficiency and autoimmunity. Autoimmun. Rev. 5(7), 465–470 (2006).
- Cunningham-Rundles C. Hematologic complications of primary immune deficiencies. Blood Rev. 16(1), 61–64 (2002).
- Ramyar A, Aghamohammadi A, Moazzami K et al. Presence of idiopathic thrombocytopenic purpura and autoimmune hemolytic anemia in the patients with common variable immunodeficiency. Iran. J. Allergy. Asthma. Immunol. 7(3), 169–175 (2008).
- Michel M, Chanet V, Galicier L et al. Autoimmune thrombocytopenic purpura and common variable immunodeficiency: analysis of 21 cases and review of the literature. Medicine (Baltimore) 83(4), 254–263 (2004).
- Horn J, Thon V, Bartonkova D et al. Anti-IgA antibodies in common variable immunodeficiency (CVID): diagnostic workup and therapeutic strategy. Clin. Immunol. 122(2), 156–162 (2007).
- Aukrust P, Lien E, Kristoffersen AK et al. Persistent activation of the tumor necrosis factor system in a subgroup of patients with common variable immunodeficiency – possible immunologic and clinical consequences. Blood 87(2), 674–681 (1996).
- Cunningham-Rundles C, Lieberman P, Hellman G, Chaganti RS. Non-Hodgkin lymphoma in common variable immunodeficiency. Am. J. Hematol. 37(2), 69–74 (1991).
- Cunningham-Rundles C. Autoimmune manifestations in common variable immunodeficiency. J. Clin. Immunol. 28(Suppl. 1), S42–S45 (2008).
- Wakim M, Shah A, Arndt PA et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am. J. Hematol. 76(2), 152–155 (2004).
- Nos P, Bastida G, Beltran B, Aguas M, Ponce J. Crohn’s disease in common variable immunodeficiency: treatment with antitumor necrosis factor alpha. Am. J. Gastroenterol. 101(9), 2165–2166 (2006).
- Daniels JA, Lederman HM, Maitra A, Montgomery EA. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): a clinicopathologic study and review. Am. J. Surg. Pathol. 31(12), 1800–1812 (2007).
- Khodadad A, Aghamohammadi A, Parvaneh N et al. Gastrointestinal manifestations in patients with common variable immunodeficiency. Dig. Dis. Sci. 52(11), 2977–2983 (2007).
- Ebrahimi Daryani N, Aghamohammadi A, Mousavi Mirkala MR et al. Gastrointestinal complications in two patients with common variable immunodeficiency. Iran. J. Allergy. Asthma. Immunol. 3(3), 149–152 (2004).
- Sneller MC. Common variable immunodeficiency. Am. J. Med. Sci. 321(1), 42–48 (2001).
- Washington K, Stenzel TT, Buckley RH, Gottfried MR. Gastrointestinal pathology in patients with common variable immunodeficiency and X-linked agammaglobulinemia. Am. J. Surg. Pathol. 20(10), 1240–1252 (1996).
- Chua I, Standish R, Lear S et al. Anti-tumour necrosis factor-alpha therapy for severe enteropathy in patients with common variable immunodeficiency (CVID). Clin. Exp. Immunol. 150(2), 306–311 (2007).
- Conlong P, Rees W, Shaffer JL et al. Primary antibody deficiency and Crohn’s disease. Postgrad. Med. J. 75(881), 161–164 (1999).
- Ravindran J, Gillis D, Rowland R, Heddle R. Common variable immunodeficiency associated with nodular regenerative hyperplasia of the liver. Aust. N. Z. J. Med. 25(6), 741 (1995).
- Malamut G, Ziol M, Suarez F et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J. Hepatol. 48(1), 74–82 (2008).
- Smith MS, Webster AD, Dhillon AP et al. Orthotopic liver transplantation for chronic hepatitis in two patients with common variable immunodeficiency. Gastroenterology 108(3), 879–884 (1995).
- Fuss IJ, Friend J, Yang Z et al. Nodular regenerative hyperplasia in common variable immunodeficiency. J. Clin. Immunol. 33(4), 348–758 (2013).
- Ward C, Lucas M, Piris J, Collier J, Chapel H. Abnormal liver function in common variable immunodeficiency disorders due to nodular regenerative hyperplasia. Clin. Exp. Immunol. 153(3), 331–337 (2008).
- Mañas MD, Marchán E, Gijón J, Martín F. Nodular regenerative hyperplasia of the liver in a patient with common variable immunodeficiency. An. Med. Interna 23(8), 395–397 (2006).
- Arenillas Rocha L, Soriano Viladomiu A, Smithson Amat A. Nodular regenerative hyperplasia and common variable immunodeficiency. Gastroenterol. Hepatol. 26(8), 524 (2003).
- Agarwal S, Cunningham-Rundles C. Autoimmunity in common variable immunodeficiency. Curr. Allergy Asthma Rep. 9(5), 347–352 (2009).
- Raeiszadeh M, Kopycinski J, Paston SJ et al. The T cell response to persistent herpes virus infections in common variable immunodeficiency. Clin. Exp. Immunol. 146(2), 234–242 (2006).
- Chua I, Quinti I, Grimbacher B. Lymphoma in common variable immunodeficiency: interplay between immune dysregulation, infection and genetics. Curr. Opin. Hematol. 15(4), 368–374 (2008).
- Abolhassani H, Aghamohammadi A, Imanzadeh A, Mohammadinejad P, Sadeghi B, Rezaei N. Malignancy phenotype in common variable immunodeficiency. J. Investig. Allergol. Clin. Immunol. 22(2), 133–134 (2012).
- Kinlen LJ, Webster AD, Bird AG et al. Prospective study of cancer in patients with hypogammaglobulinaemia. Lancet 1(8423), 263–266 (1985).
- Iglesias Alzueta J, Matamoros Florí N. Common variable immunodeficiency. Review. Allergol. Immunopathol. (Madr.) 29(3), 113–118 (2001).
- Dhalla F, da Silva SP, Lucas M, Travis S, Chapel H. Review of gastric cancer risk factors in patients with common variable immunodeficiency disorders, resulting in a proposal for a surveillance programme. Clin. Exp. Immunol. 165(1), 1–7 (2011).
- Gompels MM, Hodges E, Lock RJ et al.; Associated Study Group. Lymphoproliferative disease in antibody deficiency: a multi-centre study. Clin. Exp. Immunol. 134(2), 314–320 (2003).
- Ochtrop ML, Goldacker S, May AM et al. T and B lymphocyte abnormalities in bone marrow biopsies of common variable immunodeficiency. Blood 118(2), 309–318 (2011).
- Rizzi M, Neumann C, Fielding AK et al. Outcome of allogeneic stem cell transplantation in adults with common variable immunodeficiency. J. Allergy Clin. Immunol. 128(6), 1371–1374.e2 (2011).
A review on guidelines for management and treatment of common variable immunodeficiency
To obtain credit, you should first read the journal article. After reading the article, you should be able to answer the following, related, multiple-choice questions. To complete the questions (with a minimum 70% passing score) and earn continuing medical education (CME) credit, please go to www.medscape.org/journal/expertimmunology. Credit cannot be obtained for tests completed on paper, although you may use the worksheet below to keep a record of your answers. You must be a registered user on Medscape.org. If you are not registered on Medscape.org, please click on the New Users: Free Registration link on the left hand side of the website to register. Only one answer is correct for each question. Once you successfully answer all post-test questions you will be able to view and/or print your certificate. For questions regarding the content of this activity, contact the accredited provider, [email protected]. For technical assistance, contact [email protected]. American Medical Association's Physician's Recognition Award (AMA PRA) credits are accepted in the US as evidence of participation in CME activities. For further information on this award, please refer to http://www.ama-assn.org/ama/pub/category/2922.html. The AMA has determined that physicians not licensed in the US who participate in this CME activity are eligible for AMA PRA Category 1 Credits™. Through agreements that the AMA has made with agencies in some countries, AMA PRA credit may be acceptable as evidence of participation in CME activities. If you are not licensed in the US, please complete the questions online, print the AMA PRA CME credit certificate and present it to your national medical association for review.
Activity Evaluation: Where 1 is strongly disagree and 5 is strongly agree
1. You are seeing an 18-year-old young woman with recurrent upper respiratory and gastrointestinal infections. She was just diagnosed with common variable immunodeficiency (CVID). What can you tell her about this diagnosis?
□ A It may be diagnosed among children as young as 2 years of age
□ B The principal clinical presentation of CVID is recurrent infections
□ C Non-encapsulated bacteria promote most bacterial infections among patients with CVID
□ D Campylobacter species promote most gastrointestinal infections among patients with CVID
2. You decide to initiate immunoglobulin G (IgG) replacement therapy for this patient. What should you consider regarding this therapy?
□ A Only intravenous therapy provides consistently sufficient doses of IgG
□ B Subcutaneous doses of IgG should not exceed 100 mg/kg per week
□ C Subcutaneous administration of IgG provides a more stable serum level of IgG compared with intravenous administration
□ D Side effects are more common among adolescents vs adults over age 65
3. The patient continues to have a cough after 2 courses of antibiotic therapy. What should you consider regarding pulmonary complications of CVID?
□ A Younger age is associated with more severe pulmonary complications
□ B Her dosage of IgG may need to be increased
□ C Plain radiography remains the best means to evaluate chronic pulmonary symptoms among patients with CVID
□ D Corticosteroids are to be avoided in the management of pulmonary disease among patients with CVID
4. What else should you consider regarding the potential complications of CVID?
□ A Autoimmune thrombocytopenic purpura and autoimmune haemolytic anemia are the most common autoimmune complications of CVID
□ B CVID-related enteropathy usually improves after gluten is eliminated from the diet
□ C CVID is associated with a higher risk of hematological but not solid malignancy
□ D The most common malignancy associated with CVID is chronic leukocytic leukemia