Abstract
Multiple sclerosis is a progressive demyelinating neurological disease resulting in long-term disability, commonly manifesting in walking impairment and reduced quality of life. The use of the potassium channel blocker fampridine, chemically 4-aminopyridine, as an immediate-release formulation to improve action potential conduction in demyelinated axons was hampered by adverse events, including seizures. The prolonged-release formulation of fampridine (known as modified- or sustained-release fampridine in some countries and dalfampridine extended release in the USA) has a longer apparent half-life and a lower peak plasma concentration versus immediate-release fampridine formulations, sustaining its duration of action and reducing the incidence of adverse events. Prolonged-release fampridine is the first drug specifically licensed to improve walking in patients with multiple sclerosis, and has been shown to consistently improve walking ability in a third of patients. Prolonged-release fampridine Timed-Walk Responders showed both clinically significant improvements in walking speed and in patient-reported walking ability.
A 2.0 score indicates minimal clinical disability and fully ambulatory. A 7.0 score indicates the need for a wheelchair for mobility.
†After median times of 8, 20 and 30 years following disease onset, most patients with multiple sclerosis will reach EDSS scores of ≥4.0, ≥6.0 and >7.0, respectively Citation[3,4,7].
‡Almost 50% of patients with multiple sclerosis will reach an EDSS of 6.0 within 25 years of disease evolution and approximately 50% will reach an EDSS score of 7.0 within 30 years Citation[8].
EDSS: Expanded Disability Status Scale.
![Figure 1. Disability progression in people with multiple sclerosis as defined by the Expanded Disability Status Scale.A 2.0 score indicates minimal clinical disability and fully ambulatory. A 7.0 score indicates the need for a wheelchair for mobility.†After median times of 8, 20 and 30 years following disease onset, most patients with multiple sclerosis will reach EDSS scores of ≥4.0, ≥6.0 and >7.0, respectively Citation[3,4,7].‡Almost 50% of patients with multiple sclerosis will reach an EDSS of 6.0 within 25 years of disease evolution and approximately 50% will reach an EDSS score of 7.0 within 30 years Citation[8].EDSS: Expanded Disability Status Scale.](/cms/asset/f10cdbec-3758-46f0-9a11-69641cc325e0/ierj_a_11209539_f0001_b.jpg)
Net membrane current (inward current plotted downwards) at the start of a stretch of continuous conduction (as in a demyelinated fiber). Horizontal dashed lines indicate the zero net current. (A) Control; (B) after application of 5 mM 4-aminopyridine; (C) change in net membrane current attributable to 4-aminopyridine derived by subtracting curve B from curve A.
Reproduced with permission from Citation[27].
![Figure 3. Time course of current blocked by 4-aminopyridine (fampridine).Net membrane current (inward current plotted downwards) at the start of a stretch of continuous conduction (as in a demyelinated fiber). Horizontal dashed lines indicate the zero net current. (A) Control; (B) after application of 5 mM 4-aminopyridine; (C) change in net membrane current attributable to 4-aminopyridine derived by subtracting curve B from curve A.Reproduced with permission from Citation[27].](/cms/asset/b9b7c5fc-4f44-4fbc-ba3c-284d5eaf17ad/ierj_a_11209539_f0003_b.jpg)
(A) Significantly more patients treated with prolonged-release fampridine experience ≥10 to ≥40% improvements in walking speed on the Timed 25-Foot Walk compared with placebo. (B) Improvements in ≥20% objectively measured walking speed are associated with clinically meaningful improvements in subjectively measured walking ability.
*p < 0.05.
**p < 0.001 versus placebo.
†Measured using the Timed 25-Foot Walk.
MSWS-12: 12-item MS Walking Scale; PR: Prolonged release.
Data taken from Citation[61,62].
![Figure 4. Incremental increases in walking speed among prolonged-release fampridine- and placebo-treated patients and the association between increased walking speed and self-reported walking ability.(A) Significantly more patients treated with prolonged-release fampridine experience ≥10 to ≥40% improvements in walking speed on the Timed 25-Foot Walk compared with placebo. (B) Improvements in ≥20% objectively measured walking speed are associated with clinically meaningful improvements in subjectively measured walking ability.*p < 0.05.**p < 0.001 versus placebo.†Measured using the Timed 25-Foot Walk.MSWS-12: 12-item MS Walking Scale; PR: Prolonged release.Data taken from Citation[61,62].](/cms/asset/8568caf2-d60c-44bf-ac1e-70f0b00e279e/ierj_a_11209539_f0004_b.jpg)
DMT: Disease-modifying therapies; EDSS: Expanded Disability Status Scale; GA: Glatiramer acetate; LEMMT: Lower extremity manual muscle test; PPMS: Primary–progressive multiple sclerosis; PRMS: Progressive–relapsing multiple sclerosis; RRMS: Relapsing–remitting multiple sclerosis; SPMS: Secondary–progressive multiple sclerosis.
Data taken from Citation[69].
![Figure 5. Prolonged-release fampridine Timed-Walk Responder rate by baseline characteristics.DMT: Disease-modifying therapies; EDSS: Expanded Disability Status Scale; GA: Glatiramer acetate; LEMMT: Lower extremity manual muscle test; PPMS: Primary–progressive multiple sclerosis; PRMS: Progressive–relapsing multiple sclerosis; RRMS: Relapsing–remitting multiple sclerosis; SPMS: Secondary–progressive multiple sclerosis.Data taken from Citation[69].](/cms/asset/ff2a01c2-7ca4-4b3a-95ad-a64e52a796e0/ierj_a_11209539_f0005_b.jpg)
Introduction to multiple sclerosis
Multiple sclerosis (MS) is a progressive neurological disease resulting in long-term disability, with a median global incidence of 2.5 per 100,000 Citation[101]. The pathological hallmark of MS is inflammatory foci of demyelination in the CNS that in the short term leads to interruptions of axonal conduction and eventually may result in axonal degeneration Citation[1]. The resulting neurological deficits underlie the characteristic disabling symptoms of the disease Citation[1]. Collectively, in relapsing–remitting and secondary–progressive MS, the following negative prognostic indicators have been identified: higher initial relapse frequency, greater disability progression in the first 5 years and the involvement of cerebellar, spinal or pyramidal functional systems Citation[2]. The most influential clinical factor in reaching disability milestones is suggested to be age at clinical onset of MS: the earlier the onset, the younger the age at assignment of irreversible disability Citation[3,4]. However, recent evidence suggests that this effect is gender specific, with young males having a higher tendency for more rapid disease progression Citation[5]. As a result of its early onset and long duration, MS is associated with a disproportionate socioeconomic burden relative to more common medical conditions, such as stroke, and is estimated to cost over US$2 million/€1.6 million (US$ to € exchange rate: 0.7974 on 28 May 2012) over the lifetime of an individual Citation[6].
The general prognosis of MS is well documented, with irreversible limitation in ambulation (Expanded Disability Status Scale [EDSS] ≥4.0), a unilateral aid required for walking (EDSS ≥6.0), and patients becoming wheelchair-bound (EDSS >7.0) after median times of approximately 8, 20 and 30 years since disease onset, respectively Citation[3,7]. Natural history studies suggest that almost 50% of individuals will reach an EDSS of 6.0 within 25 years of disease evolution and approximately 50% will reach an EDSS of 7.0 within 30 years Citation[8]. With pharmacological treatment evolving, survival in MS has increased and longer times to irreversible disability have been reported in recent studies Citation[9]. Wide variation in the progression of MS is evident both within and between natural history studies, driven by differences in assessment and data collection methodology together with the heterogeneous nature of the disease itself Citation[10]. Reduced productivity and employment are suggested to be the greatest contributors to this burden, which are amplified by the early age of disease onset and long disease duration Citation[11]. Walking impairment in particular can have a profoundly negative impact on the ability to work Citation[12], the need for informal and formal care Citation[12], the ability to perform activities of daily living (ADL) Citation[13], healthcare burden Citation[14] and health-related quality of life (HRQoL) Citation[15,16]. Moreover, patients with MS have identified maintenance of mobility as a priority, and in one study of 166 patients with MS it was ranked as the most important of 13 bodily functions across all stages of the disease Citation[17].
Body of review
Overview of the market
Since the early 1990s, several pharmacological treatments with immunomodulatory or immunosuppressive properties have been developed to treat MS, commonly designated ‘disease-modifying treatments’ (DMTs). DMTs represent a major step in the control of the disease and some, such as IFN-bCitation[18], natalizumab Citation[19] and fingolimod Citation[20], have been proven to delay disability progression Citation[21]. None of the DMTs specifically address MS symptom management, and their role in ameliorating walking impairment in particular is uncertain. In the past, the symptomatic drug management of walking impairment in patients with MS relied on strategies adapted from other conditions and indications. Spasticity, for example, is a common symptom of MS and may affect walking ability. This symptom is usually addressed with oral baclofen or tizanidine Citation[22], although there are no conclusive data of the functional effects of these treatments in patients with MS Citation[23]. More recently, nabiximols has been used as an add-on therapy for those refractory to conventional antispasticity treatment Citation[24]. In March 2010, the US FDA approved a botulinum A toxin (Botox®, Allergan Inc.) for focal upper-limb spasticity in disorders including MS. While these treatments may have an effect on MS patients’ spasticity, historically no drug therapy was developed to achieve functional improvements in walking overall; nonmedicinal approaches such as physiotherapy or hippotherapy are sometimes utilized, in spite of a lack of evidence, in an attempt to achieve functional improvements.
Despite the importance of addressing spasticity in MS, it is important to recognize that many complex inter-relating factors are required for walking, such as sensory inputs, including visual control and balance, together with coordination and motor control. Walking impairment may occur due to the disruption of one or more of these sensory factors, making it a particularly difficult symptom to treat effectively. For example, balance, balance confidence, lower-extremity power and pulmonary function have all been demonstrated as significant independent determinants of walking distance in patients with MS Citation[25]. In addition, fatigue is a common debilitating symptom in patients with MS, which may manifest as exhaustion, increased drowsiness or worsening of MS symptoms Citation[26].
Blockade of potassium (K+) channels is thought to have the potential to promote nerve conduction in demyelinated neurons, and thus K+ channel blockers have been considered a potential therapeutic strategy in MS and other demyelinating diseases for over 30 years Citation[27]. However, it is only recently that this strategy has resulted in licensed treatments to improve walking impairment in individuals with MS. Fampridine, chemically 4-aminopyridine (4-AP), is a broad-spectrum K+ channel-blocking agent that has a long and varied history of applications, including use as a research tool for investigating ion channel function and for the treatment of spinal cord injury and various neurological conditions Citation[27,28]. A prolonged-release formulation of fampridine was developed in the 1990s Citation[29], and is now known as prolonged-release fampridine (or modified- or sustained-release fampridine in some markets and dalfampridine extended release in the USA), and has recently been approved by regulatory bodies in several countries as a pharmacological treatment to improve walking in people with MS.
Chemistry
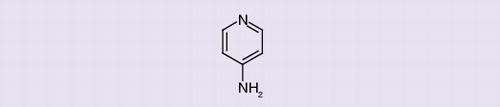
4-AP is a member of a family of mono- and di-amino derivatives of pyridine, with the molecular structure C5H6N2 and a molecular weight of 94.12 g. 4-AP can be manufactured by heating pyridine with sodium amide in N,N-dimethylaniline at 180°C Citation[102]. The compound has high aqueous solubility, based on a low octanol/water partition coefficient (LogP: -0.76) Citation[102].
Pharmacodynamics
The classical view on the mechanism of action of 4-AP is that the beneficial therapeutic effects result from blocking voltage-gated K+ (Kv) channels, which leads to prolonged depolarization and improved action potential conduction in demyelinated nerve fibers Citation[27]. In mammalian myelinated nerves, the nodes of Ranvier are densely populated with sodium (Na+) channels and sparsely populated with Kv channels Citation[30]. The fast activation and inactivation of nodal Na+ channels and the important nodal leakage current are sufficient to regenerate and repolarize the axon, without the involvement of Kv channels Citation[31]. However, in demyelinated and damaged axons, Kv channels are redistributed in a manner resembling that of Na+ channels, necessitating tight regulation of their function for axon potential generation and propagation Citation[32]. Therefore, the dose-dependent blockade of fast Kv channels by 4-AP following alterations in their expression and distribution resulting from demyelination Citation[32,33] may also help to explain its efficacy in patients with MS.
Kv channels are distributed in multiple cell types in the CNS (neurons and microglia) Citation[34]. However, it is still unclear which of the large number of Kv channels are affected by 4-AP Citation[35]. For example, in preclinical experiments, 4-AP has also been reported to potentiate synaptic neurotransmitter release at the neuromuscular junction and other synapses Citation[36], which has been suggested as a possible mechanism responsible for the beneficial action of fampridine in patients with MS. However, it is unclear how nonspecific enhancement of synaptic transmission or increases in muscle twitch tension would adequately compensate for the prevailing axonal demyelination of MS and bring about a coordinated response. Part of the uncertainty regarding the mechanism of action of 4-AP is due to its inhibitory effect on many subtypes of Kv channels, with a dose-dependent mechanism of action. In addition, the plasma concentrations of 4-AP/fampridine achieved at clinical doses are considerably lower than those used in laboratory studies to characterize its actions on various Kv channel subtypes Citation[35]. In general, the sensitivity of different Kv channels varies from micromolar to millimolar concentrations of 4-AP and within each channel the sensitivity depends on its activation state. The concentration dependence of synaptic effects has not been established in sufficient detail to help establish their putative therapeutic role in MS Citation[28].
The blockade of Kv channels may also have an impact outside of the CNS Citation[34], although the clinical implications of these changes in MS are not yet known. Since no severe adverse effects have been observed outside of the CNS at therapeutic doses, broader effects are not an immediate safety concern under therapeutic conditions. Rather, the pharmacological properties of 4-AP have prompted extensive investigation of its therapeutic potential for symptom management in disorders of neuromuscular transmission and in demyelinating diseases, particularly in MS.
Pharmacokinetics & metabolism
Many of the early clinical applications of immediate-release fampridine used intravenous delivery Citation[37], oral solutions Citation[38] or enteric coated tablets and capsules containing 4-AP Citation[37,39,40]. Capsulated immediate-release formulations have been shown to have short time to peak serum concentrations (~1.0 h) Citation[38] and an apparent plasma half-life of approximately 3.5 h Citation[38,40]. Studies of immediate-release fampridine formulations demonstrated that maximum serum concentrations (46.4 ng/ml for a 10-mg dose in fasted healthy adults) were achieved approximately 1 h after oral administration regardless of dosage Citation[38,41]. Following oral administration of 15 mg 14C-radiolabeled immediate-release fampridine, 96.4% of primarily unchanged parent compound was recovered in the urine and 0.5% in the feces after 24 h Citation[42]. Biliary excretion of immediate-release fampridine represented only a minor route of elimination Citation[42], which is concordant with the low octanol/water partition coefficient of 4-AP Citation[102]. So far, 4-amino-3-hydroxypyridine and 4-amino-3-hydroxypyridine sulfate are the only two metabolites to be detected in the urine, and they account for <10% of urinary excretion, indicating limited biotransformation Citation[42]. Negligible protein binding by immediate-release fampridine has been reported Citation[40], which is consistent with the short time to peak serum concentration and plasma half-life measured in vivo.
Although there is evidence to suggest that immediate-release fampridine may be beneficial in treating specific symptoms of MS, it has a narrow therapeutic window and may actually have convulsant activity at higher plasma concentrations Citation[43]. The CNS side effects, particularly seizure and other common adverse events (AEs) seen with immediate-release fampridine probably arise from the indiscriminate blockade of widely distributed and varied CNS Kv channels rather than Kv channels within demyelinated nerve fibers Citation[43]. The AE profile of immediate-release formulations appeared to be dose- and plasma concentration-dependent, with AEs occurring more frequently at plasma concentrations of >100 ng/ml and an intensity that correlated with the time of Cmax shortly after dosing (1–2 h) Citation[37,41]. Therefore, the authors of this research recommended clinically useful target concentrations in the range of 30–59 ng/ml Citation[41].
Pharmacokinetic studies have shown that improvement in neurological deficits is primarily related to the total 4-AP dose, while seizure induction is related to maximum plasma concentrations (Cmax) Citation[44]. Following a small randomized, double-blind, placebo-controlled, crossover study of immediate-release fampridine, conducted in 70 patients with clinically definite MS Citation[37], it was found that side effects arising 30–45 min following oral dosing could be related to high serum levels of fampridine. An open-label, follow-up extension trial subsequently reported its findings in 1994 and came to the conclusion that the value of the drug required further evaluation, given the occurrence of side effects such as seizures Citation[45]. Despite the absence of evidence-based data from large clinical trials to support its use in patients with MS, compounded immediate-release fampridine is still available from compounding pharmacies with a physician’s prescription. A recent FDA report found that at least a third of all compounded drugs fail to meet standard quality tests Citation[103]. Although not specifically mentioned in the FDA report, compounded immediate-release fampridine may be subject to these quality concerns, including variable product uniformity, potency, and contamination, which can affect a drug’s efficacy and safety. Indeed, there has been a published report of a severe accidental overdose of 4-AP due to a compounding error by a pharmacy Citation[46]. This, therefore, demonstrates the importance of standardized manufacturing processes, provided by industrial production rather than individual pharmacies, which may not have the required processes to ascertain product quality and purity.
To reduce side effects, a prolonged-release formulation of fampridine was developed, which was found to have a pharmacokinetic profile that allowed twice-daily dosing at 12-h intervals. Pharmacokinetic studies of therapeutic doses of the prolonged-release formulation have demonstrated that this formulation has an extended time to peak plasma concentration and lower peak plasma concentration relative to the immediate-release formulation Citation[47,48]. The prolonged-release formulation has approximately twice the apparent half-life (~6 h) compared with immediate-release formulations (3.7 h), while Cmax is approximately halved (~23.4 vs 46.4 ng/ml) Citation[38,47,49]. With prolonged-release fampridine, the overall drug exposure is maintained over time, as measured by the area under the concentration time curve Citation[47], whereas the time to maximum plasma concentration (Tmax) for prolonged-release fampridine is approximately tripled compared with immediate-release fampridine (3.6 vs 1.2 h) Citation[38,47,49]. In the MS-F204 Phase III clinical trial, mean plasma concentrations of prolonged-release fampridine were between 28.5 and 30.2 ng/ml at each of the double-blind visits (standard deviation: 11.2–13.3 ng/ml; range: 0–87.3 ng/ml) Citation[50]. Furthermore, at 8–10 h post-dose, the mean plasma concentration was still 21.2 ± 9.7 ng/ml Citation[50], demonstrating a sustained plasma concentration of the drug and thus the potential for maintained efficacy throughout the day.
In view of the fact that 4-AP is excreted primarily via the kidneys and with limited biotransformation, a small trial (n = 20) of prolonged-release fampridine (10 mg twice daily) was conducted in healthy volunteers and those with renal impairment Citation[47]. The mean elimination half-life was 6.4 h in healthy individuals, compared with 7.4, 8.1 and 14.3 h in patients with mild, moderate and severe renal impairment, respectively. The Cmax for fampridine, which in studies of immediate-release fampridine is the pharmacokinetic parameter linked with increased incidence of AEs Citation[37,41], was consistently higher in subjects with renal impairment following treatment with prolonged-release fampridine in comparison with healthy individuals receiving this medication Citation[47].
Regulatory aspects
Dalfampridine extended-release tablets (AMPYRA®, manufactured for Acorda Therapeutics, Inc., under license from Alkermes Pharma Ireland Limited (APIL); dalfampridine is the US adopted name of the international nonproprietary name fampridine) was first approved in 2010 in the USA, to improve walking impairment in patients with MS, as demonstrated by an increase in walking speed Citation[104]. Since then, prolonged-release fampridine (FAMPYRA®, marketing authorization held by Biogen Idec, Inc. in all markets outside of the USA; known as modified- or sustained-release fampridine in some countries), has been approved in Europe, Australia and New Zealand, and more recently in Canada . Due to its Kv channel-blocking mechanism of action, prolonged-release fampridine is contraindicated in patients with a history of seizure in all licensing countries. Moderate and severe renal impairment are also contraindications in the USA, Australia and New Zealand, as is mild renal impairment in all other licensing countries Citation[104–106]. In the EU, precaution is advised when using prolonged-release fampridine concomitantly with organic cation transporter 2 (OCT2; SLC22A2) substrates Citation[107] (e.g., carvedilol, propranolol and metformin) Citation[51]. Furthermore, the concomitant use of competitive inhibitors of organic cation transporter 2 (e.g., cimetidine) Citation[52] with prolonged-release fampridine is contraindicated Citation[107].
Clinical efficacy, safety & tolerability
Early-stage & Phase I clinical trials of immediate-release fampridine in MS
Immediate-release fampridine was first evaluated for efficacy in patients with MS in small studies conducted in the 1980s and 1990s . Dose-ranging studies using different formulations of immediate-release fampridine indicated a potential for efficacy, as improvements were noted in a number of sensory (visual function) and motor (power, coordination and gait) domains as well as improvements in muscle strength Citation[53]. Clinical utility was indicated in some patients, although concerns were raised regarding AEs, particularly seizures observed at high doses of immediate-release fampridine. Such AEs were expected given the mechanism of action of fampridine Citation[54], but it was noted that for clinical use, seizure risk would have to be assessed and minimized. Subsequent studies confirmed that the dose of fampridine affected severe AEs but not necessarily efficacy Citation[55], forming the foundation of future trials utilizing lower doses of prolonged-release fampridine Citation[50,56].
Phase II & III clinical trials of prolonged-release fampridine in MS
Phase II dose-ranging studies
The first Phase II dose-ranging trial of prolonged-release fampridine (MS-F201) evaluated the safety and efficacy of this oral tablet formulation versus placebo in patients with MS (n = 36) Citation[55]. The dose of prolonged-release fampridine was increased from 10 to 40 mg twice daily in weekly increments of 5 mg twice daily. Efficacy was assessed using a variety of measures including a modified version of the Brief Fatigue Inventory, the Modified Fatigue Impact Scale, all elements of the MS Functional Composite (MSFC) and the Lower Extremity Manual Muscle Test (LEMMT). In this trial, five patients discontinued treatment with prolonged-release fampridine owing to AEs at doses ≥25 mg twice daily, whereas no patients discontinued owing to AEs in the placebo group. The AEs leading to these treatment-related withdrawals included convulsions in two patients at doses of 30 and 35 mg twice daily. Both of the patients who experienced these convulsions made a full recovery.
No significant changes were observed in this small trial on the efficacy assessments used, which was thought to be due to a combination of AEs at the highest doses and a high degree of variability in placebo-treated patients Citation[55]. Indeed, when the post-baseline change in walking speed was adjusted for the wide variation in the Timed 25-Foot Walk (T25FW) in the placebo group in post hoc analyses, prolonged-release fampridine demonstrated a significant benefit over placebo. Similar analyses of the LEMMT data demonstrated a significant benefit on this measure. Taken together, the findings of this trial suggested that prolonged-release fampridine doses up to 20 mg twice daily should be evaluated further with lower extremity strength and walking speed as potential outcome measures.
The second trial (MS-F202) examined the efficacy and safety of three doses (10, 15 or 20 mg twice daily) of prolonged-release fampridine versus placebo in 206 patients with MS Citation[57]. The T25FW was the primary outcome measure used to determine changes in walking speed from baseline. Efficacy was also assessed utilizing the MSFC Citation[58], LEMMT, Ashworth score, the Clinician Global Impression (CGI) and Subject Global Impression (SGI), the MS Quality of Life Inventory and the patient-rated 12-item MS Walking Scale (MSWS-12) as secondary outcome measures. The initial analysis of these data revealed that walking speed on the T25FW improved from baseline by 8, 11 and 7% in the 10-, 15- and 20-mg groups, respectively, and 3% in the placebo group. However, there were no statistically significant differences in walking speed between placebo and prolonged-release fampridine at any dose. There were, however, significant treatment differences on the LEMMT for the 10 and 15 mg twice-daily doses of prolonged-release fampridine, but not for any of the other secondary measures of efficacy.
However, further examination of the data from MS-F202 identified a subset of patients (~30%) who consistently walked faster during the double-blind treatment period Citation[57]. From this, the investigators concluded that a suitable criterion for identifying a responder would be consistent improvement in walking speed during treatment relative to non-treatment periods Citation[57]. A post hoc response analysis was therefore undertaken to determine the percentage of patients who consistently had a faster walking speed on at least three out of four double-blind treatment visits compared with the maximum walking speed on any of the five off-treatment visits (four before treatment and one at 2 weeks after treatment) Citation[57]. Patients who fulfilled this criterion are defined as Timed-Walk Responders (TWRs) and those who did not meet this criterion are Timed-Walk non-Responders (TWnRs). There were significantly higher percentages of TWRs in the prolonged-release fampridine 10 mg twice daily (35.3%), 15 mg twice daily (36.0%) and 20 mg twice daily (38.6%) treatment groups than in the placebo group (8.5%). The clinical relevance of this responder criterion was assessed by comparing differences between TWRs and TWnRs on other efficacy assessments. TWRs demonstrated significantly greater improvements than TWnRs on the MSWS-12 and the SGI Citation[57].
In the MS-F202 study, severe AEs occurred at higher rates in the prolonged-release fampridine 15 and 20 mg twice-daily groups than in the 10 mg twice-daily or placebo groups. Serious AEs were also more frequent in the prolonged-release fampridine 15 and 20 mg twice-daily groups than the 10 mg twice-daily group. However, MS relapse (3.8%) and seizure (1.3%) were the only serious AEs that were reported by more than one of the patients treated with prolonged-release fampridine. Based on the safety and efficacy findings of this trial, 10 mg twice daily approximately 12 h apart was deemed to be the appropriate dose to use in future studies. As the TWR analysis used in this trial provides a sensitive and clinically relevant means of measuring treatment response to prolonged-release fampridine, this definition of response was used prospectively in the Phase III trials.
Phase III pivotal trials & extension studies
Patients & trial design
Two randomized, placebo-controlled trials (MS-F203 and MS-F204) Citation[50,56] were conducted to assess the efficacy and safety of prolonged-release fampridine 10 mg twice daily. Eligible patients had clinically definite MS of any disease course and were able to complete the T25FW in 8–45 s at the screening stage. Patients were excluded who had onset of MS exacerbation within 60 days of screening, a history of seizures and treatment with corticosteroids from 30 days prior to the screening visit. Baseline demographics and clinical characteristics for the patients included in the two Phase III studies are shown in . The median EDSS score was 6.0 (range: 1.5–7.0), indicating that the majority of patients experienced walking impairment severe enough to preclude full-day activities and require the use of walking aids for distances up to 100 m Citation[4].
Efficacy
The primary efficacy measure was based on walking speed, measured by the T25FW and performed according to the instructions of the MSFC Citation[58]. Consistent improvement on T25FW was used to define response Citation[57] in both studies, with the percent of TWRs in each treatment group as the primary efficacy measure. In both Phase III trials, prolonged-release fampridine 10 mg twice daily improved walking in patients with MS . The TWR criterion was met by 35% of patients in the prolonged-release fampridine group versus 8% in the placebo group in the MS-F203 study Citation[56] and 43% in the prolonged-release fampridine group versus 9% in the placebo group in the MS-F204 study Citation[50]. The average improvement from baseline in walking speed was 25% among TWRs in the prolonged-release fampridine group in both Phase III studies, compared with <10% for TWnRs and the placebo group . Among TWRs, increased walking speed was observed at the first post-baseline visit (following 2 weeks of treatment) and was maintained for the duration of the double-blind treatment phase in both the MS-F203 and MS-F204 trials Citation[50,56].
The MSWS-12 Citation[59] was used to assess the clinical meaningfulness of the TWR definition using the patient’s self-report of walking ability. In both trials, MSWS-12 scores changed by more than six points from baseline in TWRs and by <1 point for TWnRs . Studies aimed at determining what constitutes a clinically meaningful change on this measure suggest that the minimum clinically important difference for the MSWS-12 is between four and six points Citation[60]. The change in MSWS-12 scores reported in both clinical trials therefore represent a clinically meaningful improvement in walking ability for people with MS. Significantly more patients treated with prolonged-release fampridine experience ≥10, ≥20, ≥30 and ≥40% improvements in T25FW walking speed compared with placebo Citation[61]. In addition, incremental increases in objective walking speed measured by the T25FW are also associated with incremental improvements in self-reported walking ability on the MSWS-12 Citation[62]. These data therefore provide further evidence demonstrating that prolonged-release fampridine increases walking speed to an extent that is perceived by the patient.
Further evidence for the efficacy of prolonged-release fampridine comes from the long-term, open-label extension (LTE) studies of the two Phase III trials (MS-F203EXT and MS-F204EXT) Citation[63–65]. As expected, after prolonged-release fampridine discontinuation in the parent trials, improvement in walking speed in TWRs was lost Citation[64]. Upon treatment reinitiation in the extension studies, improvement returned at the initial assessment (2 weeks after screening), confirming the early onset of efficacy Citation[64]. In these trials, walking speed remained faster among TWRs compared with TWnRs but declined in parallel in both groups over the treatment period Citation[65]. This decline is likely to be due to deterioration in walking speed that occurs as MS progresses rather than reduced efficacy of prolonged-release fampridine or therapeutic tolerance. In support of this, a decline in walking speed of 19% (mean change: -0.5 feet/s) over 2 years has recently been reported in placebo-treated patients with MS from the Phase III IMPACT trial Citation[66,67]. Treatment benefits between prolonged-release fampridine TWRs and TWnRs were maintained for up to 2.5 years in the extension studies, indicating the potential for prolonged duration of efficacy Citation[64]. A subsequent analysis demonstrated that this benefit was maintained over approximately 3.5 years of open-label use Citation[63].
MS exhibits profound heterogeneity in clinical course, neuroradiological appearance of the lesions, disability status, concomitant DMT use and response to therapy Citation[7,68]. For this reason, it is particularly important to confirm efficacy across a range of subgroups defined by baseline disease characteristics or demographics and concomitant DMT use. A post hoc analysis of data combined from the two Phase III trials (MS F203/F204; placebo: n = 190; prolonged-release fampridine TWRs: n = 129; prolonged-release fampridine TWnRs: n = 214) reporting efficacy in such groups has recently become available Citation[69]. Subgroup analyses demonstrated that the percentage of prolonged-release fampridine TWRs was similar for patients with relapsing versus progressive courses of MS and was independent of disease duration . TWR rates were also similar in those with differing degrees of ambulatory disability at baseline as measured by EDSS ≤5.5, 6.0 or ≥6.5 . Furthermore, concomitant use of DMTs such as IFN-𝛃 or glatiramer acetate did not significantly alter TWR rates . The percentage of prolonged-release fampridine TWRs was 31% when spasticity and muscle weakness were present (Ashworth score >1 and LEMMT score ≤4) and 39% when these symptoms were absent (Ashworth score ≤1 or LEMMT score >4) .
The magnitude of improvement in walking speed for TWRs as measured by percentage and absolute change in walking speed (m/s) or walking time (s) from baseline was also similar across all subgroups. These empirical improvements were supported by patient-reported subjective improvements on the MSWS-12. Mean change MSWS-12 scores showed a consistent improvement (decrease) across subgroups in prolonged-release fampridine TWRs (range: -2.49 to -10.54) versus placebo (range: 3.33 to -1.99) and prolonged-release fampridine TWnRs (range: 3.00 to -2.67). Interestingly, approximately a third of patients receiving prolonged-release fampridine treatment were identified as TWRs across all subgroups, which corresponds to the responder rate reported in the original two Phase III trials Citation[50,56]. These data confirm that baseline MS disease characteristics or concomitant use of DMTs have little impact on the efficacy of prolonged-release fampridine and signify its suitability for treating walking impairment across a diverse range of patients with MS.
Safety
In both Phase III trials, prolonged-release fampridine was generally well tolerated and AE profiles were similar Citation[50,56]. The percentage of patients in the prolonged-release fampridine treatment group experiencing any AE was 84 and 86% in the MS-F203 and MS-F204 studies, respectively, whereas the percentage of placebo patients who reported AEs was 81 and 66%, respectively . AEs that occurred in >5% of the prolonged-release fampridine-treated groups in the MS-F203 and MS-F204 trials are listed in , together with AEs that occurred in at least 2% of patients and more frequently with prolonged-release fampridine versus placebo across the entire clinical development program. In the MS-F203 study, one patient in the prolonged-release fampridine group had a focal seizure following treatment of sepsis secondary to community-acquired pneumonia. No seizures occurred in the prolonged-release fampridine group in the MS-F204 study but one complex, partial seizure occurred in the placebo group and led to withdrawal from the trial.
The long-term safety and tolerability of prolonged-release fampridine was further assessed in the LTE studies Citation[64,65]. In both trials, all patients received prolonged-release fampridine at 10 mg twice daily approximately 12 h apart, with assessments at 2, 14 and 26 weeks and then every 6 months. There was at least a 2-week washout period at the end of the double-blind studies, during which time study treatment was stopped and was reinitiated upon extension trial enrolment Citation[63]. Only data for patients randomized to prolonged-release fampridine treatment in the MS-F203/204 trials and who completed at least one T25FW evaluation were reported Citation[63]. Tolerability was consistent during both open-label LTE trials and no new safety signals emerged Citation[50,56,63]. In the extension studies, only four seizure-related AEs were reported (0.8%) Citation[65], which is comparable to the incidence rates noted in a comprehensive review of seizure risk in patients with MS Citation[70].
Postmarketing safety
Safety data are now available from the exposure of approximately 62,400 patients in the USA to dalfampridine extended release (35,000 patient-years) from product approval (January 2010) through March 2012 Citation[71]. The most frequently reported postmarketing AEs were similar to those most often seen during clinical development, including dizziness, insomnia, balance disorder, headache, nausea, urinary tract infection, back pain and asthenia Citation[71]. As part of the risk evaluation and mitigation strategy required by the FDA, data on incidences of seizures and convulsions were specifically elicited. A total of 160 seizures were either reported or confirmed by a healthcare practitioner (~4.6/1000 patient-years of use), as of 31 March 2012. Of the patients who experienced seizures, more than 54% had an additional potential risk factor for seizure, such as use of concomitant medications with a labeled seizure risk (n = 17), a previous history of seizure (n = 10), incorrect dosing (n = 8), a prior head injury (n = 1) or renal impairment (n = 5).
The safety and tolerability of prolonged-release fampridine in MS over an extended period (longer than 12 months) is currently under investigation in the Canadian Interventional Extension Trial (ClinicalTrials.gov identifier: NCT01235221 Citation[109]) and its use in routine clinical practice is being investigated in the observational LIBERATE trial (ClinicalTrials.gov identifier: NCT01480063 Citation[109]). Data are estimated to be available in 2013 and 2014, respectively, and are expected to provide valuable insights on the long-term use of prolonged-release fampridine in patients with MS. Furthermore, data regarding HRQoL assessed using the physical component scale of the short-form 36 (SF36) health questionnaire should also become available from the currently active ENABLE trial (ClinicalTrials.gov identifier: NCT01480076 Citation[109]) in 2013.
Conclusion
Prolonged-release fampridine consistently improves walking speed in a third of patients with MS and is generally well tolerated. As the only drug specifically licensed to improve walking in MS, prolonged-release fampridine provides the best defense against walking disability, which is common and regarded as highly debilitating by both physicians and their patients. Prolonged-release fampridine has been proven to be efficacious in the same proportion of patients with MS regardless of their baseline demographics or disease characteristics, or their concomitant use of DMTs, thus signifying its suitability for treating walking-related symptoms across a diverse range of patients with MS. Patients who respond to prolonged-release fampridine have been demonstrated to show clinically significant improvements in walking speed and in patient-reported walking ability. Based on the association between increased walking speed and increases in HRQoL Citation[13,15], it is hoped that prolonged-release fampridine therapy may help to improve HRQoL, enhance the undertaking of ADL and reduce healthcare costs.
Expert commentary & five-year view
One key area for future research is the assessment of the feasibility and utility of novel walking assessments and the clinical meaningfulness of change on these novel measures. Candidate end points include the Berg Balance Scale Citation[72] and the Timed Up and Go test Citation[72]. Further research on the physiological changes of impaired gait associated with prolonged-release fampridine treatment may provide additional insight into how this can be improved. The impact of prolonged-release fampridine on MS patients’ HRQoL is of particular interest, as data have shown that declines in walking speed are associated with declines in HRQoL and ADL in MS patients Citation[13]. The Phase IV ENABLE trial (ClinicalTrials.gov identifier: NCT01480076 Citation[109]) is currently investigating the relationship between response to prolonged-release fampridine and HRQoL.
The success of prolonged-release fampridine in treating walking impairment in MS is likely to drive research into its possible efficacy in treating other symptoms of MS or other neurological diseases characterized by demyelination or conduction block. A search of the clinical trials database in February 2012 identified several ongoing trials to assess the use of prolonged-release fampridine for the treatment of various MS symptoms Citation[109]. In addition to the LIBERATE and ENABLE trials described above, trials assessing the dosing of prolonged-release fampridine in MS (ClinicalTrials.gov identifier: NCT01328379 Citation[109]), as well as trials in patients with optic neuritis (ClinicalTrials.gov identifier: NCT01337986 Citation[109]), cerebral palsy (ClinicalTrials.gov identifier: NCT01468350 Citation[109]) and Parkinson’s disease (ClinicalTrials.gov identifier: NCT01491022 Citation[109]) are either planned or underway. Given the proposed mechanism of action of prolonged-release fampridine and initial evidence from the clinical use of this medication, the drug may have other beneficial effects on other CNS-mediated MS symptoms, such as optic neuritis and manual function.
Table 1. Existing drug labels of fampridine across the world.
Table 2. Early clinical trials of immediate-release fampridine (4-aminopyridine) for the treatment of multiple sclerosis.
Table 3. Demographics and baseline characteristics in two Phase III trials (MS-F203 and MS-F204).
Table 4. The efficacy of prolonged-release fampridine as assessed in two Phase III trials (MS-F203 and MS-F204).
Table 5. Adverse events summary from the MS-F203 and MS-F204 trials, and combined analyses of the most common adverse events observed in the clinical trials of prolonged-release fampridine 10 mg twice daily.
Key issues
• Walking impairment is one of the most common and feared disabling features of multiple sclerosis (MS), which contributes to reduce health-related quality of life and increased healthcare burden.
• Prolonged-release fampridine, chemically 4-aminopyridine, is a broad-spectrum potassium channel-blocking agent that promotes nerve transmission in demyelinated neurons.
• Prolonged-release fampridine is the first MS drug specifically licensed to improve walking in people with MS.
• Therapeutic use of immediate-release formulations of fampridine was hampered by a poor pharmacokinetics profile, responsible for a high incidence of adverse events and an unacceptable rate of seizures.
• Compared with the immediate-release formulation, prolonged-release fampridine has a longer apparent plasma half-life, which contributes to its sustained duration of action, and a lower peak plasma concentration, which reduces the incidence of adverse events to placebo-like levels.
• Prolonged-release fampridine is generally well tolerated and exhibits a postmarketing seizure risk rate comparable to that observed in general MS populations.
• Use of prolonged-release fampridine for the treatment of other MS symptoms or as therapy in similar neurological diseases is of future interest.
Financial & competing interests disclosure
O Fernandez has received honoraria for serving as a consultant in advisory boards, or chair or speaker in meetings; and for participation in clinical trials and other research projects funded by Biogen Idec, Bayer Schering, Merck Serono, Teva and Novartis. T Berger has participated in meetings sponsored by and received honoraria (lectures, advisory boards or consultations) from pharmaceutical companies marketing treatments for multiple sclerosis: Allergan, AOP, Baxter, Bayer Schering, Biogen Idec, Biotest, CSL Behring, Merck Serono, Novartis, Sanofi and Teva. T Berger and his institution have received financial support by unrestricted research grants (Allergan, AOP, Biogen Idec, Berlex, Bayer, Biotest, CSL Behring, Merck Serono and Sanofi) and for participation in clinical trials in multiple sclerosis sponsored by Bayer Schering, Biogen Idec, Merck Serono, Novartis, Octapharma, Roche, Sanofi and Teva. H-P Hartung has received honoraria for consulting, membership of steering committees and advisory board activities from Bayer Pharmaceuticals Corporation, Biogen Idec, BioMS, Genzyme, Merck Serono, Novartis, Roche, Sanofi and Teva. N Putzki is a full-time employee of Biogen Idec. The authors received no compensation related to the development of the manuscript. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing support was provided by Samantha Forster of UBC Scientific Solutions, which was supported by Biogen Idec.
References
- Dutta R, Trapp BD. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog. Neurobiol. 93(1), 1–12 (2011).
- Degenhardt A, Ramagopalan SV, Scalfari A, Ebers GC. Clinical prognostic factors in multiple sclerosis: a natural history review. Nat. Rev. Neurol. 5(12), 672–682 (2009).
- Confavreux C, Vukusic S. Age at disability milestones in multiple sclerosis. Brain 129(Pt 3), 595–605 (2006).
- Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an Expanded Disability Status Scale (EDSS). Neurology 33(11), 1444–1452 (1983).
- Scalfari A, Neuhaus M, Daumer M et al. Long-term evolution of ‘benign’ multiple sclerosis patients in the London Ontario Database [P01.138]. Presented at: The 64th Annual Meeting of the American Academy of Neurology. LA, USA, 21–28 April 2012.
- Whetten-Goldstein K, Sloan FA, Goldstein LB, Kulas ED. A comprehensive assessment of the cost of multiple sclerosis in the United States. Mult. Scler. 4(5), 419–425 (1998).
- Confavreux C, Vukusic S, Adeleine P. Early clinical predictors and progression of irreversible disability in multiple sclerosis: an amnesic process. Brain 126(Pt 4), 770–782 (2003).
- Motl RW, McAuley E. Association between change in physical activity and short-term disability progression in multiple sclerosis. J. Rehabil. Med. 43(4), 305–310 (2011).
- Kingwell E, van der Kop M, Zhao Y et al. Relative mortality and survival in multiple sclerosis: findings from British Columbia, Canada. J. Neurol. Neurosurg. Psychiatr. 83(1), 61–66 (2012).
- Tremlett H, Zhao Y, Rieckmann P, Hutchinson M. New perspectives in the natural history of multiple sclerosis. Neurology 74(24), 2004–2015 (2010).
- Jennum P, Wanscher B, Frederiksen J, Kjellberg J. The socioeconomic consequences of multiple sclerosis: a controlled national study. Eur. Neuropsychopharmacol. 22(1), 36–43 (2012).
- Piercy J, Rajagopalan K, Pike J et al. Burden of walking problems in MS: analysis of caregiver and indirect costs. Neurology 76(9), A606 (2011).
- Salter AR, Cutter GR, Tyry T, Marrie RA, Vollmer T. Impact of loss of mobility on instrumental activities of daily living and socioeconomic status in patients with MS. Curr. Med. Res. Opin. 26(2), 493–500 (2010).
- Larocca NG. Impact of walking impairment in multiple sclerosis: perspectives of patients and care partners. Patient 4(3), 189–201 (2011).
- Polman CH, Havrdova E, Confavreux C et al. Relationship between Timed 25-Foot Walk walking speed and health-related quality of life in AFFIRM and SENTINEL (#1250). Presented at: The 64th Annual Meeting of the American Association of Neurology. LA, USA, 21–28 April 2012.
- Jones CA, Pohar SL, Warren S, Turpin KV, Warren KG. The burden of multiple sclerosis: a community health survey. Health Qual. Life Outcomes 6, 1 (2008).
- Heesen C, Böhm J, Reich C, Kasper J, Goebel M, Gold SM. Patient perception of bodily functions in multiple sclerosis: gait and visual function are the most valuable. Mult. Scler. 14(7), 988–991 (2008).
- Oliver BJ, Kohli E, Kasper LH. Interferon therapy in relapsing–remitting multiple sclerosis: a systematic review and meta-analysis of the comparative trials. J. Neurol. Sci. 302(1), 96–105 (2011).
- Polman CH, O’Connor PW, Havrdova E et al.; AFFIRM Investigators. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 354(9), 899–910 (2006).
- Kappos L, Radue EW, O’Connor P et al.; FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 362(5), 387–401 (2010).
- Berger JR. Functional improvement and symptom management in multiple sclerosis: clinical efficacy of current therapies. Am. J. Manag. Care 17(Suppl. 5), S146–S153 (2011).
- Paisley S, Beard S, Hunn A, Wight J. Clinical effectiveness of oral treatments for spasticity in multiple sclerosis: a systematic review. Mult. Scler. 8(4), 319–329 (2002).
- Shakespeare DT, Boggild M, Young C. Anti-spasticity agents for multiple sclerosis. Cochrane Database Syst. Rev. 4, CD001332 (2003).
- Novotna A, Mares J, Ratcliffe S et al.; Sativex Spasticity Study Group. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex®), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur. J. Neurol. 18(9), 1122–1131 (2011).
- Wetzel JL, Fry DK, Pfalzer LA. Six-minute walk test for persons with mild or moderate disability from multiple sclerosis: performance and explanatory factors. Physiother. Can. 63(2), 166–180 (2011).
- Vucic S, Burke D, Kiernan MC. Fatigue in multiple sclerosis: mechanisms and management. Clin. Neurophysiol. 121(6), 809–817 (2010).
- Bostock H, Sears TA, Sherratt RM. The effects of 4-aminopyridine and tetraethylammonium ions on normal and demyelinated mammalian nerve fibres. J. Physiol. (Lond.) 313, 301–315 (1981).
- Hayes KC. The use of 4-aminopyridine (fampridine) in demyelinating disorders. CNS Drug Rev. 10(4), 295–316 (2004).
- Schwid SR, Petrie MD, McDermott MP, Tierney DS, Mason DH, Goodman AD. Quantitative assessment of sustained-release 4-aminopyridine for symptomatic treatment of multiple sclerosis. Neurology 48(4), 817–821 (1997).
- Compston A, Coles A. Multiple sclerosis. Lancet 359(9313), 1221–1231 (2002).
- Schwarz JR, Reid G, Bostock H. Action potentials and membrane currents in the human node of Ranvier. Pflugers Arch. 430(2), 283–292 (1995).
- Karimi-Abdolrezaee S, Eftekharpour E, Fehlings MG. Temporal and spatial patterns of Kv1.1 and Kv1.2 protein and gene expression in spinal cord white matter after acute and chronic spinal cord injury in rats: implications for axonal pathophysiology after neurotrauma. Eur. J. Neurosci. 19(3), 577–589 (2004).
- Fehlings MG, Nashmi R. Changes in pharmacological sensitivity of the spinal cord to potassium channel blockers following acute spinal cord injury. Brain Res. 736(1-2), 135–145 (1996).
- Espejo C, Montalban X. Dalfampridine in multiple sclerosis: from symptomatic treatment to immunomodulation. Clin. Immunol. 142(1), 84–92 (2012).
- Dunn J, Blight A. Dalfampridine: a brief review of its mechanism of action and efficacy as a treatment to improve walking in patients with multiple sclerosis. Curr. Med. Res. Opin. 27(7), 1415–1423 (2011).
- Wu ZZ, Li DP, Chen SR, Pan HL. Aminopyridines potentiate synaptic and neuromuscular transmission by targeting the voltage-activated calcium channel beta subunit. J. Biol. Chem. 284(52), 36453–36461 (2009).
- Van Diemen HA, Polman CH, Koetsier JC, Van Loenen AC, Nauta JJ, Bertelsmann FW. 4-aminopyridine in patients with multiple sclerosis: dosage and serum level related to efficacy and safety. Clin. Neuropharmacol. 16(3), 195–204 (1993).
- Hayes KC, Katz MA, Devane JG et al. Pharmacokinetics of an immediate-release oral formulation of fampridine (4-aminopyridine) in normal subjects and patients with spinal cord injury. J. Clin. Pharmacol. 43(4), 379–385 (2003).
- Donnelly R. Chemical stability of 4-aminopyridine capsules. Can. J. Hosp. Pharm. 57, 283–287 (2004).
- Uges DR, Sohn YJ, Greijdanus B, Scaf AH, Agoston S. 4-aminopyridine kinetics. Clin. Pharmacol. Ther. 31(5), 587–593 (1982).
- Bever CT Jr, Young D, Anderson PA et al. The effects of 4-aminopyridine in multiple sclerosis patients: results of a randomized, placebo-controlled, double-blind, concentration-controlled, crossover trial. Neurology 44(6), 1054–1059 (1994).
- Blight AR, Henney HR 3rd. Pharmacokinetics of 14C-radioactivity after oral intake of a single dose of 14C-labeled fampridine (4-aminopyridine) in healthy volunteers. Clin. Ther. 31(2), 328–335 (2009).
- Judge SI, Lee JM, Bever CT Jr, Hoffman PM. Voltage-gated potassium channels in multiple sclerosis: overview and new implications for treatment of central nervous system inflammation and degeneration. J. Rehabil. Res. Dev. 43(1), 111–122 (2006).
- Bever CT, Judge SI. Sustained-release fampridine for multiple sclerosis. Expert Opin. Investig. Drugs 18(7), 1013–1024 (2009).
- Polman CH, Bertelsmann FW, van Loenen AC, Koetsier JC. 4-aminopyridine in the treatment of patients with multiple sclerosis. Long-term efficacy and safety. Arch. Neurol. 51(3), 292–296 (1994).
- Schwam E. Severe accidental overdose of 4-aminopyridine due to a compounding pharmacy error. J. Emerg. Med. 41(1), 51–54 (2011).
- Smith W, Swan S, Marbury T, Henney H 3rd. Single-dose pharmacokinetics of sustained-release fampridine (fampridine-SR) in healthy volunteers and adults with renal impairment. J. Clin. Pharmacol. 50(2), 151–159 (2010).
- Vollmer T, Blight AR, Henney HR 3rd. Steady-state pharmacokinetics and tolerability of orally administered fampridine sustained-release 10-mg tablets in patients with multiple sclerosis: a 2-week, open-label, follow-up study. Clin. Ther. 31(10), 2215–2223 (2009).
- Vollmer T, Henney HR 3rd. Pharmacokinetics and tolerability of single escalating doses of fampridine sustained-release tablets in patients with multiple sclerosis: a Phase I–II, open-label trial. Clin. Ther. 31(10), 2206–2214 (2009).
- Goodman AD, Brown TR, Edwards KR et al.; MSF204 Investigators. A Phase 3 trial of extended release oral dalfampridine in multiple sclerosis. Ann. Neurol. 68(4), 494–502 (2010).
- Zolk O, Solbach TF, König J, Fromm MF. Functional characterization of the human organic cation transporter 2 variant p.270Ala>Ser. Drug Metab. Dispos. 37(6), 1312–1318 (2009).
- Kido Y, Matsson P, Giacomini KM. Profiling of a prescription drug library for potential renal drug–drug interactions mediated by the organic cation transporter 2. J. Med. Chem. 54(13), 4548–4558 (2011).
- Davis FA, Stefoski D, Rush J. Orally administered 4-aminopyridine improves clinical signs in multiple sclerosis. Ann. Neurol. 27(2), 186–192 (1990).
- Jones RE, Heron JR, Foster DH, Snelgar RS, Mason RJ. Effects of 4-aminopyridine in patients with multiple sclerosis. J. Neurol. Sci. 60(3), 353–362 (1983).
- Goodman AD, Cohen JA, Cross A et al. Fampridine-SR in multiple sclerosis: a randomized, double-blind, placebo-controlled, dose-ranging study. Mult. Scler. 13(3), 357–368 (2007).
- Goodman AD, Brown TR, Krupp LB et al.; Fampridine MS-F203 Investigators. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet 373(9665), 732–738 (2009).
- Goodman AD, Brown TR, Cohen JA et al.; Fampridine MS-F202 Study Group. Dose comparison trial of sustained-release fampridine in multiple sclerosis. Neurology 71(15), 1134–1141 (2008).
- Fischer JS, Rudick RA, Cutter GR, Reingold SC. The Multiple Sclerosis Functional Composite Measure (MSFC): an integrated approach to MS clinical outcome assessment. National MS Society Clinical Outcomes Assessment Task Force. Mult. Scler. 5(4), 244–250 (1999).
- Hobart JC, Riazi A, Lamping DL, Fitzpatrick R, Thompson AJ. Measuring the impact of MS on walking ability: the 12-Item MS Walking Scale (MSWS-12). Neurology 60(1), 31–36 (2003).
- Hobart JC. Prolonged-release fampridine for multiple sclerosis: was the effect on walking ability clinically significant (P509). Mult. Scler. 16, S7–S39, S172 (2010).
- Limmroth V, Putzki N, Goodman AD. Data from prolonged-release fampridine trials confirm that 20% improvement in walking speed is clinically meaningful. Presented at: The 27th Congress of the European Committee for Treatment and Research in Multiple Sclerosis. Amsterdam, The Netherlands, 19–22 October 2011.
- Hobart JC, Blight AR, Lynn F et al. The Timed 25-Foot Walk as an outcome measure for clinical trials in multiple sclerosis: further evidence that a 20% change is clinically meaningful. Presented at: The 21st Meeting of the European Neurological Society. Lisbon, Portugal, 28–31 May 2011.
- Goodman AD. Updated analysis of open-label extension studies of dalfampridine extended release tablets in multiple sclerosis. Presented at: The 27th Congress of the European Committee for Treatment and Research in Multiple Sclerosis. Amsterdam, The Netherlands, 19–22 October 2011.
- Goodman AD, Brown TR, Edwards KR et al. Analysis of open-label extension studies of prolonged release fampridine tablets in multiple sclerosis. Mult. Scler. 16, S175 (2010).
- Goodman AD, Blight A. Final Data from Open-label extension studies of dalfampridine extended release tablets in multiple sclerosis [P04.128]. Presented at: The 64th Annual Meeting of the American Academy of Neurology. LA, USA, 21–28 April 2012.
- Cohen JA, Cutter GR, Fischer JS et al.; IMPACT Investigators. Benefit of INF-𝛃-1a on MSFC progression in secondary progressive MS. Neurology 59(5), 679–687 (2002).
- Krishnan A, Goodman AD, Potts J et al. Health-related quality of life is reduced in multiple sclerosis patients whose walking speed declines over time. Presented at: The 64th Annual Meeting of the American Association of Neurology. LA, USA, 21–28 April 2012.
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47(6), 707–717 (2000).
- Stourac P, Putzki N. Prolonged-release fampridine shows consistent efficacy across subgroups of multiple sclerosis patients. Presented at: The 22nd meeting of the European Neurological Society. Prague, Czech Republic, 9–12 June 2012.
- Koch C, Uyttenboogaart M, Polman S, De Keyser J. Seizures in multiple sclerosis. Epilepsia 49(6), 948–958 (2008).
- Jara M, Adera M, Adedeji A, Henney HR 3rd, Carrazana EJ. Dalfampridine extended release tablets: safety profile after 2 years of postmarketing experience in the United States. Presented at: The 28th Congress of the European Committee for Treatment and Research in Multiple Sclerosis. Lyon, France, 10–13 October 2012.
- Cattaneo D, Regola A, Meotti M. Validity of six balance disorders scales in persons with multiple sclerosis. Disabil. Rehabil. 28(12), 789–795 (2006).
- Stefoski D, Davis FA, Faut M, Schauf CL. 4-aminopyridine improves clinical signs in multiple sclerosis. Ann. Neurol. 21(1), 71–77 (1987).
- Stefoski D, Davis FA, Fitzsimmons WE, Luskin SS, Rush J, Parkhurst GW. 4-aminopyridine in multiple sclerosis: prolonged administration. Neurology 41(9), 1344–1348 (1991).
- van Diemen HA, Polman CH, van Dongen TM et al. The effect of 4-aminopyridine on clinical signs in multiple sclerosis: a randomized, placebo-controlled, double-blind, cross-over study. Ann. Neurol. 32(2), 123–130 (1992).
Websites
- WHO and The Multiple Sclerosis International Federation. Atlas multiple sclerosis resources in the world, 2008. www.who.int/entity/mental_health/neurology/Atlas_MS_WEB.pdf (Accessed 21 June 2012)
- Hazardous Substances Data Bank and TOXNET. 4-aminopyridine, 2012. http://toxnet.nlm.nih.gov/cgi-bin/sis/search/r?dbs+hsdb:@term+@rn+@rel+504-24-5 (Accessed 13 June 2012)
- US FDA. 2006 limited FDA survey of compounded drug products. www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/PharmacyCompounding/ucm204237.htm (Accessed 21 June 2012)
- Acorda Therapeutics, Inc. AMPYRA® [dalfampridine] extended release tablets prescribing information, 2010. http://ampyra.com/home (Accessed 21 June 2012)
- EMA. FAMPYRA: summary of product characteristics, 2011. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002097/WC500109956.pdf (Accessed 17 October 2012)
- Australian Government, Department of Health and Ageing, Therapeutic Goods Administration. Australian Public Assessment Report for fampridine, 2012. www.tga.gov.au/auspar/auspar-fampridine-110615.htm (Accessed 17 October 2012)
- European Medical Agency. EPAR. Summary of opinion (initial authorisation) for FAMPYRA (fampridine), 2011. www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002097/human_med_001432.jsp&mid=WC0b01ac058001d124 (Accessed 30 November 2011)
- Health Canada. Drugs and healthcare products, 2012. www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2012_fampyra_132859-eng.php (Accessed 17 October 2012)
- ClinicalTrials.gov. http://clinicaltrials.gov