Abstract
A substantial body of evidence supports a role for the growth hormone (GH)–IGF-1 axis in cancer incidence and progression. This includes epidemiological evidence relating elevated plasma IGF-1 to cancer incidence as well as a lack of cancers in GH/IGF-1 deficiency. Rodent models lacking GH or its receptor are strikingly resistant to the induction of a wide range of cancers, and treatment with the GH antagonist pegvisomant slows tumor progression. While GH receptor expression is elevated in many cancers, autocrine GH is present in several types, and overexpression of autocrine GH can induce cell transformation. While the mechanism of autocrine action is not clear, it does involve both STAT5 and STAT3 activation, and probably nuclear translocation of the GH receptor. Development of a more potent GH receptor antagonist or secretion inhibitor is warranted for cancer therapy.
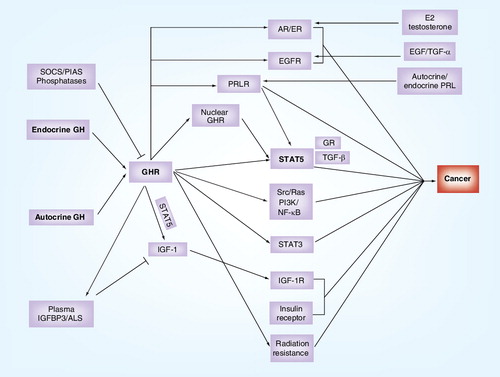
GH receptor activation by endocrine or autocrine GH is feedback regulated by SOCS, phosphatases and PIAS. Receptor activation induces transcription of IGF-1, and the receptors for prolactin, EGF, estrogen and androgen, as well as promoting insulin synthesis and secretion. While GH receptor activation results in activation of oncogenic Src, Ras/ERK, PI3-kinase and NF-κB pathways, it also activates oncogenic STATs, STAT3 and particularly STAT5. The latter complexes with glucocorticoid receptors and TGF-β, and is necessary for the transforming actions of nuclear localized GHR. Finally, IGF-1 produced as a result of GHR activation and regulated by GH-dependent IGFBP3/ALS is a key element in tumor promotion. Text in bold shows standard abbreviaitons.
ALS: Acid labile subunit; AR: Androgen receptor; EGFR: EGF receptor; ER: Estrogen receptor; GH: Growth hormone; GHR: Growth hormone receptor; GR: Glucocorticoid receptor; IGFBP3: Insulin-like growth factor-binding protein 3; PIAS: Protein inhibitors of activated signal transducer and activator of transcription; PRL: Pituitary hormone prolactin; PRLR: Pituitary hormone prolactin receptor; SOCS: Suppressor of cytokine signaling proteins; STAT: Signal transducer and activator of transcription.
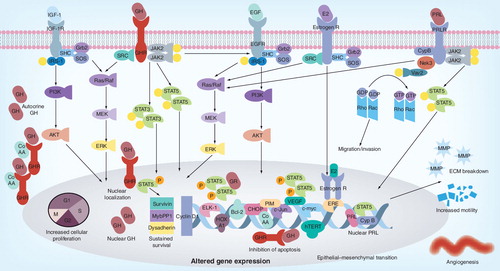
GH and PRL signaling via Jak2 mediate many of the downstream responses through phosphorylation of STAT transcription factors, MAP kinases and other kinase cascades. STAT5 and STAT3 are major contributors to the downstream signaling and upregulate key proliferative genes. In PRLR-mediated signaling the phosphorylation of Vav2 (a guanosine nucleotide exchange factor that stimulates the Rho/Rac protein family) by Nek3 allows it to enhance the activity of Rac responsible for enhanced motility and invasion. The somatotrophic effects of GH are mediated by IGF-1 and its receptor (IGF-1R) serves as a potent proliferative signaling system that stimulates cell growth and promotes cell survival. The proliferative actions of IGF-1 are mediated by the association of receptor tyrosine kinase with SHC, Grb2 and SOS that activate the Ras and MAPK cascade (Raf, Mek and Erk1/2). This cascade culminates in activation of Elk-1 and other transcription factors. The anti-apoptotic effects are mediated by the phosphorylation of IRS-1 via the PI3K pathway resulting in the activation of Akt and mTOR. A cell exhibiting upregulation of these key signaling molecules or lack of a negative feedback has increased mitogenic potential with inhibition of apoptotic pathways. GHR nuclear localization, a function of highly proliferative cells, can promote in tumorigensis via the Jak2/STAT5 pathway together with CoAA, dependent on autocrine GH. This results in dysregulation of expression of survivin, dysadherin and Mybbp1a – genes associated with tumorigenesis. PRL can also be translocated into the nucleus by binding to CypB, which enhances the STAT5 DNA-binding activity and prolactin-induced STAT5-mediated gene expression. Autocrine GH increases cell proliferation (via HoxA1, TFF3, Jak/STAT, MAPK, Cyclin D1, c-myc, hTERT), survival (via TFF3, Bcl2, CHOP, Jak/STAT) and migration/invasion/EMT (via TFF3, MMP, γ-catenin) and angiogenesis (TFF3, VEGF) by differential regulation of bracketed genes. estrogen receptor, androgen receptor and EGFR upregulated by GH signaling also contributes to tumor promotion by genomic and plasma membrane actions.
ECM: Extracellular matrix; GH: Growth hormone; GHR: Growth hormone receptor; PRL: Prolactin.
Six decades of research have established that growth hormone (GH) plays a crucial role in promoting proportionate postnatal growth, and that this is largely mediated through the induction of IGF-1. This action is supported by important roles of GH in the regulation of metabolism, and of cardiovascular, renal, reproductive and immune functions. These roles are accomplished through ubiquitously expressed GH receptors (GHRs) which, similar to receptors for many other growth factors, signal through a tyrosine kinase (JAK2) able to activate STAT, Ras/ERK and PI3K/Akt pathways. The ability of GH to also induce IGF-1, both locally in a paracrine/autocrine manner Citation[1,2] and in an endocrine manner via action on the liver, results in a particularly potent growth stimulus that requires tight control. This comprises feedback inhibition of GH secretion by pituitary somatotropes by hepatic (endocrine) IGF-1, as well as a set of negative regulators common to other growth factor/cytokine signaling factors: suppressors of cytokine signaling (SOCS), protein inhibitors of activated STAT (PIAS) proteins, phosphatases, receptor downregulation, and transcriptional regulation of receptor expression. Given the ability of GH and IGF-1 to promote cell proliferation, cell movement and angiogenesis, and to suppress apoptosis, it is not surprising that dysregulation of the tightly controlled GH–IGF-1 axis promotes neoplasia. As discussed here, converging data from recent epidemiologic, animal and in vitro studies indicate that the state of the GH–IGF-1 axis has important influences on cancer biology, cancer risk and carcinogenesis.
Actions of the GH–IGF-1 axis in relation to cancer
The risk of developing cancer is determined by genetic elements and by environmental conditions including diet and lifestyle. Recent evidence suggests that the GH–IGF-1 axis might provide a link between these factors and the disposition to cancer through its effects on normal cell proliferation, differentiation and apoptosis. The GH–IGF-1 axis influences several stages and aspects of cancer development and behaviour: cellular proliferation, cell survival, angiogenesis and metastasis, and even resistance to chemotherapy Citation[3]. Being a potent and ubiquitous mitogen, IGF-1 can act on many cell types to bring about increased proliferation via the MAPK signaling pathway. Its anti-apoptotic actions are mediated via the PI3K/Akt pathway and can disrupt the tight control between cell proliferation and death leading to hyper-proliferation Citation[4]. Carcinogenesis is a multistep process which involves accumulation of a number of genomic ‘hits’ leading to complete transformation and/or survival of partially transformed cells with one or a few hits Citation[5]. In such a case, IGF-1 would increase the pool of transformed/damaged cells that are available for undergoing subsequent hits. This implies that higher levels of IGF-1 can bring about increased proliferation and survival and hence promote carcinogenesis even though not being directly responsible for initiating cancer development. Hence, individuals with relatively high levels of IGF-1 may be at an increased risk as the anti-apoptotic actions of IGF supports partially transformed cells to provide an increased pool of cells for subsequent damaging hits. It is important to note that the IGF-1 activity in a given tissue is not merely a function of its circulating levels but also the local expression of genes encoding for IGFs, IGF-1 receptor (IGF-1R), IGFBPs and proteases that cleave the IGFBPs to regulate the release of IGF-1 (particularly for IGFBP-3). There is considerable heterogeneity in the levels of IGF-1 and IGFBP-3 between normal individuals as a result of genetic and non-genetic determinants Citation[6,7].
Recent evidence suggests a complex cross-talk between the GH–IGF-1 axis and estrogen receptor signaling that can stimulate mammary epithelial proliferation under normal conditions and increase the risk of breast cancer Citation[8], as well as positive crosstalk with the ErbB1 receptor Citation[9]. The latter also undergoes direct tyrosine phosphorylation by the GHR Citation[10], which itself is able to activate the classic oncogenic pathways directly through JAK2 and Src Citation[11]. Thus, the combination of GH and IGF-1 signaling, together with their interactions with other potentially oncogenic signaling proteins such as estrogen receptor and ErbB1 would lead one to expect involvement in cancer initiation or progression for this key growth-promoting axis .
Epidemiological studies
Numerous epidemiological studies have provided evidence that GH/IGF-1 is likely to be a driver of major human cancers. First, there is a link between tall stature, cancer risk and GH/IGF-1 levels. With the use of final height as a marker for GH and IGF-1 action, a review of over 300 case–control and cohort studies Citation[12] found that taller individuals have a higher incidence of breast cancer (22% increase), prostate cancer (20% increase) and colon cancer (20–60% increase) relative to shorter individuals. A Danish study of 14-year-old children in the top quintile of height showed that they have an adjusted relative risk of over 1.5 for breast cancer, which surpasses other risk factors examined including BMI and age of first menarche Citation[13]. A carefully controlled twin study from Scandinavia also found that tallness associates with increased breast cancer risk Citation[14]. An anthropometric analysis revealed an association between rapid childhood growth during adolescence with the risk of developing several cancers, particularly of the breast, prostate and colon Citation[3]. For breast cancer risk, a major prospective meta-analysis of patients in 12 countries concluded that not only are serum IGF-1 concentrations positively associated with height, but they are also positively associated with breast cancer risk in women (high vs low serum IGF-1; OR: 1.28), but only in estrogen receptor-positive tumors Citation[15]. Moreover, higher levels of free and total plasma IGF-1 have been reported in breast cancer patients Citation[16]. In relation to the restriction of risk to estrogen receptor-positive women, synergistic interaction between estrogen action and IGF-1 action has been shown in vitro for epithelial cell proliferation Citation[17]. IGF-1 levels are also positively related to high mammographic density, which is an indicator of increased breast cancer risk Citation[18]. Of further relevance, loss of BRCA1 function associates with overactivation of the IGF-1 signaling axis Citation[19].
There have been reports that genetic variants in the GH–IGF system can determine relative tumor risk. A genome-wide association study (GWAS) in UK Caucasians has identified 64 SNPs that influence susceptibility for lung cancer, of which 11 were mapped to genes of the GH–IGF-1 axis, including GH1, GHR, GH-releasing hormone (GHRH) and IGFBP5 Citation[20], and genetic variation at many genes in the GH–IGF pathway have been shown to associate with a variety of cancers Citation[20,21]. A recent GWAS of over 1000 breast cancer cases matched with a similar number of controls concluded that the GH signaling pathway was the third most highly enriched pathway in breast cancer, but found insulin signaling intermediates rather than IGF-1 intermediates to co-associate with GH/JAK2 signaling in this analysis Citation[22]. However, polymorphisms in the IGF-1 gene or in the GH synthesis pathway were not significantly associated with breast cancer Citation[23]. Four IGFBP-3 SNPs have been reported to be associated with IGF-1 and IGFBP-3 levels in one study Citation[24], and a strong association between a specific BP-3 promoter polymorphism and mammographic density, a known risk factor for breast cancer development, in another study Citation[25]. However a larger cohort and a recent multi-ethnic cohort study reported no direct association of BP3 with breast cancer Citation[23,26]. While IGFBP-3 polymorphic alleles were modestly associated with risk of colorectal cancer Citation[27], there were contrasting associations between IGFBP-3 polymorphisms and the risk of prostate cancer in other studies Citation[28,29]. It remains to be seen if these genetic variations could determine a higher cancer risk in susceptible individuals receiving GH treatment.
Elevated levels of IGF-1 have also been shown to confer an increased risk for other cancers such as colorectal cancer and prostate cancer. A recent systematic review of 42 published studies concluded that raised circulating IGF-1 is positively associated with prostate cancer risk, with little evidence for a role of IGF-2, IGFBP-1, IGFBP-2, and inconclusive evidence for involvement of IGFBP-3 Citation[30]. For colon cancer, the relative risk for IGF-1 (RR: 1.07; 95% CI: 1.01–1.14) is modest compared with that seen in acromegaly Citation[31]. This may be a consequence of the elevated insulin levels associated with acromegaly acting together with elevated IGF-1 and, of course, GH itself Citation[31–33].
Acromegaly & cancer risk
Acromegaly is an important disease state for assessment of cancer risk because of the presence of persistently elevated levels of IGF-1. This is a result of hypersecretion of GH by pituitary somatotropes, particularly as a result of activating mutations in the GHRH signaling cascade. A number of studies have been carried out to assess whether this condition is associated with cancer risk. The most recent meta-analysis by Renehan and Brennan across three population studies concluded that acromegaly is associated with a 2.46-fold (95% CI: 1.79–3.38) increased risk of colon cancer Citation[34]. Adenomatous lesions tend to be larger and more dysplastic in acromegaly, and are often multiple Citation[35]. Acromegaly was also associated with an increased risk for thyroid cancer (3.64-fold; 95% CI: 1.63–8.11) in this meta-analysis. However, there is no statistical association between serum IGF-1 and presence of colorectal neoplasia in the three existing studies Citation[34], so one is left with the possibility of direct GH action on the colon, or elevated plasma insulin as a driving factor, consequent to the diabetogenic action of elevated GH Citation[36]. In relation to the other cancers, which showed a relationship to circulating IGF-1 in large population studies, in acromegaly there has been no confirmed association with breast cancer, although the limited sample number of untreated female acromegalics may have prevented small effects being observed Citation[35]. This could also be a consequence of impaired ovulation in acromegalic women; hence, a lack of progesterone and estrogen, which together can promote breast cancer. Likewise, prostatic cancer shows no evident association with acromegaly, although benign prostatic hyperplasia with calcifications is evident across different studies Citation[35]. A number of other cancers have been reported to be associated with acromegaly, but insufficient numbers are available for definite conclusions. These include various lympho-hematopoietic neoplasms (e.g., lymphoma, multiple myeloma, chronic myeloid or lymphocytic leukemia), brain tumors including meningiomas, adrenal tumors and melanomas Citation[35].
These studies indicate a low but significant risk for certain cancers, particularly of epithelial origin, in relation to elevated circulating IGF-1. What of the converse situation, that is, GH and/or IGF-1 deficiency? In the sole published study to date, a worldwide survey of 222 individuals with GHR mutation or isolated GH deficiency, not a single case of malignancy was reported. Conversely, 338 first- and second-degree relatives reported a 10–24% incidence of a range of malignancies Citation[37]. The difference in average age between the two groups (32 vs 55 years, respectively) probably contributed to this disparity, but the apparent resistance of GH-deficient and/or IGF-1-deficient individuals to cancer is supported by extensive animal studies. These will be discussed in the following sections, but at face value, the above findings imply that IGF-1–GH signaling is permissive for cancer but that beyond its essential role at low IGF-1 concentrations, has only modest effects in cancer promotion.
Children treated with GH & cancer risk
The administration of recombinant human GH was approved for the treatment of children with GH deficiency in 1985 and GH was one of the first therapeutic drugs to be produced by recombinant DNA technology. Over the past few years, several concerns have been raised in regard to its diabetogenic effects, adverse effects on skeletal growth, or increased risk of new or recurrent cancers Citation[38]. A recent report by Bell et al. recorded findings generated from a study of 54,996 children treated with recombinant GH, corresponding to nearly 195,000 patient-years of monitoring Citation[39]. This study found very low adverse effects of GH administration and no additional cancer cases in children without other risk factors for malignancy. However, in children with hypothyroidism, the risk of second neoplasm was increased, suggesting that GH could aggravate neoplastic tendencies resulting from underlying genetic abnormalities or radiotherapy. Another independent study by Ergun-Longmire et al. had reported the same conclusion whereby GH therapy doubled the risk of second neoplasm in childhood cancer survivors Citation[40]. Generally, most studies report that replacing circulating GH in humans does not promote cancer. However, while there were no additional cases relative to the general population, there was no data comparing GH-treated with GH-deficient untreated children for ethical reasons. Given the finding of no malignancies in the aforementioned GH/IGF-1 deficiency referred to above, one would predict that such a comparison would show an increase in cancer incidence on GH replacement to normal levels. This would be concordant with the many animal studies verifying the resistance of GH/IGF-1-deficient animals to cancer.
Animal models
The prospect that GH is an etiologic factor in tumorigenesis was evident from the early clinical observation that hypophysectomy inhibited metastasis and progression of specific malignancies Citation[41], and that high doses of GH induced neoplasms in rats Citation[42]. Supporting these early observations, it was noted that transgenic mice overexpressing human GH exhibited higher incidences of tumors in mammary glands Citation[43,44] and livers Citation[45]. These mice have constitutively activated hepatic STAT3, Src, ERK and Akt Citation[46]. An intermediatory role of IGF-1 in many GH actions is concordant with the observation that overexpression of IGF-1 in the mammary epithelium leads to development of mammary cancers Citation[47,48]. Similarly, expression of constitutively active IGF-1R leads to early development of mammary and salivary gland adenocarcinomas Citation[48]. In GH-transgenic mice crossed with SOCS-2-knockout mice, loss of the GH signaling antagonist SOCS-2 was found to be associated with increased local IGF-1 production and multiple hyperplastic and lymphoid polyps Citation[49].
Conversely, GHR-knockout mice display a lower and delayed incidence of neoplastic disease with substantially fewer fatal neoplastic lesions, particularly lymphomas and pulmonary adenoma/carcinomas compared with wild-type littermates Citation[50]. Thus, 83% of C57Bl6 littermate controls died naturally from neoplastic diseases, whereas only 42% of the GHR-deleted mice died from neoplasms in this study. In animal models of carcinogenesis, such as mammary tumors driven by the Simian virus 40 large T antigen, tumors in GHR-deleted mice are also markedly reduced in size (50 vs 776 mm3) and number (3.2 ± 1.2 vs 9.8 + 1.4) compared with those observed in the wild-type mice Citation[51]. Likewise, lit/lit mice that are characterized by a nonfunctional GHRH receptor with GH and IGF-1 levels less than 10% of wild-type counterparts, exhibit marked retardation of human MCF-7 breast cancer xenograft growth (average: 35 large tumors in wild-type vs five in lit/lit by day 39 after implantation Citation[52]). GH-deficient dwarf rats (dw/dw with 20% normal serum IGF-1) are resistant to mammary tumors induced by the carcinogen nitrosomethylurea (tumor incidence 100% in wild-type vs 4.8% in dw/dw rats) with an average tumor load of 7.0 per wild-type rat vs 1.0 per dw/dw rat. Hormone replacement of dw/dw rats with estrogen, progesterone, GH or IGF-1 revealed a similar tumor incidence and tumor burden to wild-type rats only with bGH replacement, and a lesser increase with IGF-1 replacement (62% incidence with an average of 1.6 tumors per rat), Citation[53]. This study was confirmed by Shen et al. with the additional finding that halting GH replacement led to tumor regression Citation[54]. Likewise, development of prostate carcinomas in the Probasin/TAg rat is markedly decreased on a dw/dw background such that by 52 weeks of age, the majority of dw/dw rats did not develop carcinomas, while all control Probasin/TAg rats did. Indeed, by 10 weeks of age, the dorsal and ventral prostate showed 100% tumor incidence in normal rats, while the GH-deficient dw/dw rats did not have measurable tumors Citation[55].
It is clinically relevant that mice transgenic for the GH antagonist G120R GH (with 46% normal serum IGF-1) are partially resistant to 7,12-dimethylbenz(a)anthracene (DMBA)-induced mammary tumors (31.6% free of tumors vs 68% tumor free for the antagonist mice at 39 weeks after treatment) Citation[56]. Accordingly, treatment of mice harboring MCF-7 xenografts with large doses of pegvisomant (an improved clinical version of the G120R GH antagonist) resulted in 70–80% suppression of circulating IGF-1 and a 30% decrease in tumor volume together with a twofold reduction in proliferation and a twofold increase in apoptosis. However, no effect on IGF-1-independent MDA-MB235 or MDA-MB231 xenografts was evident Citation[57]. Regression of all 15 human meningioma xenografts in nude mice was induced by daily pegvisomant treatment over 8 weeks at 45 mg/kg. Interestingly, this was accompanied by only a 20% decrease in serum IGF-1 Citation[58], suggesting a direct action of GH on this cancer. A similar study with colorectal cancer-205 xenografts by Dagnaes-Hansen et al. showed a marked decrease in tumor weight (∼40%) and increased apoptosis with second daily administration of 60 mg/kg pegvisomant, accompanied by a decrease in serum IGF-1 to 36% of the control value Citation[59].
These differences in tumor progression might relate to the extent of IGF-1 availability, as IGF-1 has been shown to act in vitro as a tumor promoter by increasing proliferation and decreasing apoptosis, and by promoting angiogenesis and metastasis Citation[4]. Indeed, local expression of IGF-1 in mouse mammary tissue resulted in spontaneous mammary tumors in 30% of mice Citation[60]. Alternatively, these differences might also relate to GH availability or to a combination of both GH and IGF-1 availability. Some conclusions can be drawn from mouse models with lower levels of circulating IGF-1 but which have normal/elevated tissue expression as a result of selective knockout of hepatic IGF-1 expression (liver IGF-1-deleted or LID mice). These mice have approximately 50% of normal circulating IGF-1, which results in a 40% significant decline in azoxymethane-induced colon adenocarcinoma number, largely as a result of increased apoptosis Citation[61]. An earlier study with orthotopically transplanted colon-38 adenocarcinomas found the circulating IGF-1 in their LID mice to be approximately 25% of normal, and this was associated with a substantial decrease in the number of mice with palpable colon tumors from 57 to 31%, together with a significant increase in tumor latency from 23 to 27 days Citation[62]. Hepatic metastases were also significantly decreased from 44 to 31%, and these improvements were reversed with IGF-1 administration. Generation of mammary tumors by chemical or genetic means is also decreased in LID mice Citation[63]. Both the DMBA carcinogen model and the C3(1)/Simian virus 40 large T-antigen expressing mouse model show decreased incidence and onset in LID mice. The decrease for DMBA-treated mice is from 56 to 26% incidence, and latency from 60 to 74 days, while with the large T-antigen model approximately 60% of mice developed one tumor compared with approximately 40% with two tumors in control mice. At 4 weeks, the tumor volume in the large T-antigen model was approximately 50% of the control mice volume. These modest differences contrast with the striking blockade to tumor development seen in the GHR-deleted mice and the GH-deficient rats and mice discussed previously. This could be a result of insufficiently low circulating IGF-1 in the LID mice, or of paracrine IGF-1 generation in response to activation of the ubiquitous GHR. The elevated circulating GH consequent to lowered circulating IGF-1 in the LID mice would not appear to be making a major contribution to cancer promotion, since the LID mice do have a significantly lower tumor burden.
These studies clearly indicate the involvement of endocrine GH and IGF-1 in tumor growth promotion, and indicate that effective therapeutic options for cancer treatment need to drastically lower serum IGF-1.
GHR expression & cancer
The GHR is expressed in almost all cells, and its expression is subject to control by nine alternate first exons in humans Citation[64]. The GHR transcript has been found to be overexpressed in a limited range of cancers, notably prostate carcinoma, glioblastoma, neuroectodermal tumors, adult T-cell lymphoma, kidney clear cell carcinoma and parathyroid adenoma (see ) Citation[201]. In addition, immunohistochemical and quantitative PCR studies have reported GHR overexpression in a high proportion of invasive breast ductal carcinoma Citation[65], adrenal cortical neoplasms Citation[66] and in colorectal cancer Citation[67]. Moreover, elevated serum GH-binding protein (derived from the GHR by cleavage, and a measure of GHR expression Citation[68]) is associated with a threefold increase in breast cancer incidence Citation[69]. Immunohistochemical assessment has also revealed GHR overexpression in melanoma Citation[70] and prostate carcinoma Citation[71]. This expression has been found to be not only cytoplasmic but also within the nucleus (see later). To date, there are no reports of constitutively active GHR mutations.
Autocrine GH & cancer
15 years ago, expression of GH was reported in the mammary tissue of dogs in response to progesterone administration, resulting in acromegalic symptoms Citation[72]. The hormone is synthesized in the hyperplastic ductular epithelium of the mammary gland, since GH mRNA was present in dog and cat epithelia, and was shown to be elevated by progesterone Citation[73]. As recently reported by Illera’s group, expression of GH protein in canine mammary tissue is markedly (20-fold) elevated in malignant tumors, and less so in benign tumors and dysplasias Citation[74]. This expression correlates with a 12-fold increase in IGF-1 protein expression and a fourfold increase in estradiol content of the malignant tumors, with a correspondingly lower but still elevated expression of IGF-1 and estradiol in benign tumors Citation[74]. Increased mammary GH and IGF-1 correlated with increased serum GH and IGF-1, with serum levels of GH greater than 6 ng/ml and IGF-1 greater than 43 ng/ml resulting in death of the majority of dogs within 6 months from neoplasia Citation[13]. Is this striking evidence for autocrine/paracrine actions of GH in cancer promotion evident in humans?
The study of Mol et al. reported the presence of GH transcripts in human mammary tissue but with no evident relation to presence of carcinogenesis, and apparently decreased transcript in most carcinomas Citation[73]. Raccurt et al. later reported increased GH protein in three intraductal carcinoma samples relative to normal tissue, but not in invasive ductal carcinomas Citation[75]. They also provided ISH and qPCR evidence for GH transcript expression in these three carcinomas. Recently, Ratkaj et al. used IHC to study human GH expression in a larger series (n = 40) of ductal invasive breast carcinomas and fibroadenomas, and concluded that GH was expressed in all fibroadenomas and their stroma, but in only 55% of ductal carcinomas and 20% of their stromal tissue Citation[76]. There was no correlation between GH expression and tumor size or tumor grade. A search of the Oncomine database reveals underexpression of GH1 and no listing for GH2 transcripts in invasive ductal breast carcinoma against normal tissue, in contrast to the prolactin transcript, which is highly expressed Citation[201]. In a survey of GH transcript expression across breast cancer lines, van Garderen and Schalken found only three of ten lines expressed GH1 transcript by PCR Citation[77], (including MCF-7 cells, confirmed by Gil-Puig Citation[78]), although all express GHR transcripts. Thus, it appears that humans are unlike dogs in overexpression of paracrine/autocrine GH in mammary cancer, but rather rely on prolactin.
What about other cancers?
Many human prostate cancers overexpress GH1 or GH2 and GHR transcripts as evidenced in the Oncomine database Citation[201]. Furthermore, an immunohistochemical study of 20 prostate cancers and 17 controls reported a fourfold increase in human GH expression in prostatic carcinoma Gleason scores 6–8. Co-expression of GH and GHR isoforms has also been reported at protein and mRNA levels in prostate cancer cell lines (e.g., ALVA-41, PC-3, DU 145) Citation[71,79], with van Garderen and Schalken reporting six of seven prostate cancer lines expressing GH1 transcripts, and all expressing GHR transcripts Citation[77]. Association of autocrine GH expression with prostate cancer contrasts with clinical studies show that acromegaly is associated with prostate hypertrophy but not malignancy Citation[80], and the observation that GH replacement in GHD patients increases prostate size to normal without any increase in neoplasia Citation[81]. As previously noted, absence of GH or GHR signaling results in resistance to T-antigen induced prostate cancer in mice and rats Citation[55,82]. Loss of GH/IGF1 activity is also associated with impaired development of the prostatic ductal branching architecture that results from IGF-1 deficiency Citation[83]. A reasonable hypothesis to explain these findings is that autocrine GH acts in a different manner from endocrine GH to promote cancer formation, whereas endocrine GH promotes hypertrophy in an IGF-1-dependent manner. This situation may apply to other cancers overexpressing GH1 and GH2 evident in the Oncomine database, such as chronic adult T-cell leukemia, ovarian mucinous carcinoma, endometrioid and clear-cell adenocarcinomas, and bladder urothelial cancers Citation[201]. The GHR itself is also reported as overexpressed in a range of neoplasias (see earlier). Moreover, since human GH can activate the human prolactin receptor Citation[84], the presence of overexpressed prolactin receptor in prostate cancer, T-cell leukemias and ovarian adenocarcinomas Citation[201] raises the possibility that autocrine GH may also drive oncogenesis in these cancers.
The oncogenic actions of forced autocrine expression of human GH in immortalized human breast lines has been extensively studied by Lobie’s group. Thus, Zhu et al. have reported that overexpression of autocrine GH in mammary epithelial cell lines (particularly MCF-7 cells) results in increased cell proliferation, transformation and invasion, with an epithelial to mesenchymal transition Citation[85]. This is associated with an altered transcript expression profile, with a number of oncogenic genes being upregulated by autocrine GH overexpression Citation[86]. Autocrine GH led to increased expression of the oncogenic homeobox protein Hox-A1 and telomerase protein together with downregulation of junctional plakoglobin and increased DNA methyltransferase-3 activity resulting in methylation of the plakoglobin promoter Citation[87]. Human GH overexpression also resulted in increased activity of matrix metalloproteases-2 and -9, redistribution of E-cadherin to the cytoplasm and increased secretion of trefoil factor 3 (TFF3), proposed to mediate the oncogenic actions of autocrine GH by Lobie’s group by paracrine actions on adjacent cells Citation[87]. Autocrine production of human GH was associated with increased angiogensis in xenografts, in accord with its ability to recruit endothelial cell precursors and to induce VEGF Citation[88]. Of interest, autocrine human GH-driven proliferation of MCF-7 cells, transcriptional activation and cell spreading was reported to be completely blocked by the addition of exogenous human GHR-specific antagonist B2036/pegvisomant Citation[89]. These studies indicate that overexpression of human GH in transformed cells is oncogenic. Is there, then, a difference between autocrine and endocrine GH signaling? van den Eijnden and Strous examined this question in ts20 cells, finding that GHR signaling in cells with autocrine GH does not manifest before the Golgi, and is associated with a very low level of chronic STAT5 and GHR tyrosine phosphorylation, but robust induction of a STAT5-responsive luciferase reporter Citation[90]. Sustained GHR signaling would be associated with induction of negative-feedback regulators such as SOCS proteins, hence the pulsatile nature of pituitary GH secretion. It would appear that sustained autocrine GH stimulation does something different, for example, results in the gain of a phenotype characteristic of oncogenic transformation, yet sustained endocrine GH stimulation (as in acromegaly) results in only a modest increase in cancer susceptibility. One is driven to the conclusion that intracellular signaling events are important in the transformation process, particularly since Strous’ group found that autocrine GH-expressing cells are insensitive to external (endocrine) GH Citation[90]. Conversely, Lobie’s group have reported that exogenous B2036/pegvisomant blocks autocrine growth, implying that autocrine GH is secreted and acts externally (or that the antagonist is internalized at high concentrations). It may be that both a hormone-induced signal from the plasma membrane and an intracellular signal are necessary for full autocrine action by GH.
Nuclear GHR & cancer
A likely possibility for altered intracellular signaling relates to the presence of the GHR in the cell nucleus, since this increases cell sensitivity to STAT5 Citation[91]. Tissue immunohistochemistry reveals that the nuclear GHR is present in many proliferating cells, including a variety of cancers, such as breast cancer Citation[92], colorectal carcinoma Citation[93], hepatocellular carcinoma Citation[94], melanoma Citation[70] and uterine cervical neoplasms Citation[95]. The translocation of a transmembrane receptor into the nucleus is known to occur for the EGFR, the FGF receptor and IFN-g receptor Citation[96], and is often associated with cell transformation. Full-length GHR is translocated to the nucleus in a GH- and importin-β-dependent manner in cell models, and in vivo in response to regeneration after partial hepatectomy in the rat, which is a GH-dependent process Citation[91]. There is a high correlation between the nuclear GHR and cell proliferation during the liver regeneration process and targeting the GHR to the nucleus by fusion with a nuclear localization signal markedly increased the cell proliferative response to autocrine GH via STAT5 activation in pro-B cells, resulting in constitutively activated STAT5. Moreover, pro-B BaF3 cell lines stably expressing nuclear-targeted GHR become transformed, and form solid tumors in immunocompromised mice. This is associated with upregulation of transcripts for a number of genes involved in oncogenesis such as survivin, dysadherin and MybBP. Accordingly, a high proportion of nuclear GHR cells are evident in sections of highly proliferative lymphomas Citation[91]. Further examination of the mechanism involved led to the identification of an important RNA splicing protein and transcriptional co-activator, CoAA, which binds to the extracellular domain of the receptor in response to GHs, potentially facilitating nuclear translocation by means of its nuclear localization motif Citation[97]. Transfection of pro-B cells with CoAA markedly enhanced the proliferative response to GHs, but not to IL-3, the physiological cytokine for these pro-B cells. CoAA is an oncogene related to Ewing’s sarcoma protein and translocated in liposarcoma (TLS) oncoproteins, which is amplified in lymphoma, non-small cell carcinoma and squamous cell carcinoma Citation[98], and can drive osteosarcoma proliferation in vitroCitation[99]. It is also a potent co-activator of the estrogen receptor, which is highly relevant to breast cancer Citation[100].
GH & prolactin receptor activation in cancer
As mentioned previously, human GH also activates the human prolactin receptor Citation[84], although lower species have GHs and prolactins specific for their cognate receptors. PRL can upregulate local IGF-1 expression in breast cancer cells, resulting in downstream activation of Erk1/2 and Akt Citation[101], and PRL and IGF-1 augment neoplastic progression synergistically Citation[102]. Following the initial observation that human GH-transgenic mice develop mammary cancer (human GH activates both GH and prolactin receptors in mice) Citation[103], Wennbo et al. created rat prolactin-transgenic and bovine GH-transgenic mice, which target their prolactin and GHRs, respectively Citation[104]. Only in the prolactin transgenics were mammary tumors evident, supporting the oncogenic role of autocrine/paracrine prolactin in mammary tumorigenesis. This may be a consequence of the differences in level of receptor expression for GH and prolactin in the mammary gland since both receptors have similar signaling pathways. The role of prolactin in breast cancer has been extensively reviewed by Clevenger et al.Citation[105]. Clinical and animal model studies also support a role for autocrine prolactin in prostatic cancer progression Citation[106,107] and elevated prolactin receptor expression is evident in colorectal carcinoma and metastases Citation[108]. These findings can be accommodated in a model of abnormal signaling by these two closely related class-1 cytokine receptors involving excessive activation, particularly of STAT5, but also of STAT3 Citation[109–111].
The role of STAT5 activation in cancer
Constitutively activated STAT5 can act as an oncogene, and is a common feature of hematopoietic proliferative diseases. This is a consequence of activating mutations in the JAK kinase or relevant cytokine receptor such as JAK V617F Citation[13,112,113], TEL-JAK2 Citation[114], Bcr-abl Citation[115] and MPL W515L Citation[116]. Activation of STAT5 has been shown to be critical for transformation by several of these oncogenic mutations Citation[117–119]. There is now accumulating evidence that activated STAT5 plays a vital role in promoting a broad range of cancers, including prostate cancer Citation[120], melanoma Citation[121,122], hepatocellular carcinoma Citation[123] and breast cancer Citation[124]. Activation of STAT5 has also been found to increase tumor growth and aggressiveness of hepatocellular carcinoma and squamous epithelial cell carcinoma by induction of epithelial–mesenchymal transition Citation[123,125]. Induction of senescence is a common feature of oncogenes Citation[126] and expression of constitutively active STAT5 has also been shown to induce senescence Citation[127,128].
Several studies investigating human melanoma patient samples and melanoma cell lines have shown that phosphorylated STAT5 is a common feature in melanoma, particularly in metastasis Citation[82,121,122]. Knockdown of STAT5 by RNAi in melanoma cells reduced Bcl-2 and cyclinD2 transcription, reduced cell viability and induced apoptosis, and expression of a dominant negative (C-terminal truncated) STAT5 inhibited expression of Bcl-2, leading to G1 arrest and enhanced cell death Citation[121,122]. In a fish model of melanoma, constitutive activation of STAT5 has shown to correlate with the aggressiveness of the melanoma Citation[129], suggesting that activation of STAT5 plays an important role in metastasis. In a 3D skin reconstruction model of metastatic melanoma, differentiation and invasion is inhibited by roscovitine Citation[130], which is an inhibitor of cyclin-dependent kinases Citation[131] and STAT5 activity Citation[132].
Similarly, activated STAT5 has been detected in the majority of prostate cancer specimens, particularly in high histological grade cancers, but not in normal prostate epithelium Citation[99,133]. It has recently been shown that activated STAT5 promotes growth and metastatic behavior of human prostate cancer cells in vitro and in vivoCitation[120,134–136] and expression of a dominant-negative STAT5 induces cell death in both androgen-independent and androgen-sensitive prostate cancer cell lines Citation[133]. Interestingly, STAT5 is able to synergise with androgen receptor by upregulating androgen receptor expression, which results in upregulation of STAT5 expression. The liganded androgen receptor and STAT5 interact in prostate cancer cells and enhance each other’s nuclear uptake Citation[137]. Because both the GHR and the prolactin receptor activate STAT5 as a key signaling pathway, it is likely that both autocrine/paracrine and endocrine GH and prolactin action would enhance active (oncogenic) STAT5 generation.
Many studies have shown STAT5 to be a major player in breast cancer Citation[124], for example, expression of dominant-negative STAT5A in T47D breast cancer cells induces apoptosis Citation[138], however, STAT5A and STAT5B show distinct effects in human mammary carcinoma cell lines Citation[110]. Transgenic mice expressing a constitutively active variant of STAT5 (a C-terminal 44 amino acid deleted STAT5 fused to the JAK2 kinase domain via residues 677–847 of STAT6) specifically in mammary epithelium via the β-lactoglobulin regulatory sequences are predisposed to mammary tumor formation Citation[139]. However, it is interesting to note that overexpression of the C-terminal deleted dominant-negative form of STAT5 from the same regulatory sequences also predisposes mice to mammary tumors; although the tumors from activated STAT5 expression showed a more differentiated phenotype suggesting different mechanisms are at play Citation[140].
To date there has been no identification of a constitutively active mutation of GHR, however, with current whole genome sequencing regimes of large numbers of tumor samples it is likely that such mutations may be identified, similar to those identified for other class-1 cytokine receptors such as the thrombopoietin receptor W515L/K Citation[141] and S505N Citation[142] activating mutations, and the constitutively active prolactin receptor found in benign tumors Citation[143].
Expert commentary
It is clear that GH/IGF-1 is necessary for the progression of a wide range of cancers in rodents and dogs. However, the situation is not so clear with humans, since only one study has reported on cancer incidence in GHR/GH-deficient patients. This needs to be rectified with further clinical data.
The relationship between autocrine GH expression and cancer incidence/progression needs to be firmly established with comprehensive studies of clinical samples.
The basis for the transforming actions of autocrine GH needs to be fully explored using cells with modest levels of GH expression corresponding to those evident in particular cancers. Prostate carcinoma would be a good model for these studies, because both autocrine GH and highly expressed GHR are present in prostate cancers.
There is no significant risk above that of the general population resulting from therapeutic replacement with GH. However, use of GH for prolonged periods at supraphysiologic levels could contribute to increased cancer risk, particularly colon and thyroid cancer, as it does in acromegaly.
Five-year view
Given the animal and human data supporting critical involvement of the GH–IGF-1 axis in cancer incidence and progression, the development of GH antagonists for use alone or in conjunction with IGF-1R blockers, is of key importance. While pegvisomant is partially effective in suppressing cancer in animal models, it is disadvantaged by high cost and likely impairment of its action by elevated plasma GH resulting from lowered IGF-1 feedback. Therefore, other means of suppressing GH secretion or action would be most useful. Nevertheless, clinical trials of pegvisomant in cancer in conjunction with other adjuvant therapies appear warranted. The ability of GH and IGF-1 to confer resistance to radiotherapy Citation[144,145] and chemotherapy Citation[146] is a further reason for use of GH antagonists.
Given the potential role of GH in promoting stem cell activation Citation[136,147], we can expect to see studies relating GH action to cancer stem cells and GH in the future.
Finally, while no activating clinical mutations in the GHR have been identified to date, this is a reasonable probability based on other activating mutations in cytokine receptors. Screening for activating mutations in GH/IGF-1-dependent cancers such as prostate cancer and lymphoma would appear warranted.
Table 1. Animal models implicating growth hormone/IGF-1 in cancer incidence and progression.
Table 2. Summary of oncomine data for overexpression of growth hormone 1, growth hormone 2 and growth hormone receptor transcript expression (top 10% or better) compared with normal tissue.
Key issues
• Epidemiological studies implicate increased growth hormone (GH)–IGF-1 axis activity in promotion of certain cancers (e.g., prostate, colon and breast cancer).
• Deficiency of endocrine GH or deletion of the GH receptor confers resistance to a wide range of cancers in rodents and humans.
• The GH antagonist pegvisomant has efficacy in slowing tumor development in animal models, including human xenografts.
• GH receptors are overexpressed in a range of cancers.
• Autocrine/paracrine GH may contribute substantially to cancer promotion in humans, as it does in dogs.
• The transforming activity of autocrine GH appears to involve sensitization to STAT5 and potentially STAT3 action.
• Both GH and prolactin can contribute to cancer progression since they both activate STAT5 and STAT3, and autocrine GH can activate the prolactin receptor in humans.
• GH and IGF-1 will synergise to promote cancer cell proliferation, survival and metastasis.
Financial & competing interests disclosure
Michael J Waters is supported by grants from the National Health and Medical Research Council (Australia). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Related Research Data
References
- Nilsson A, Carlsson B, Isgaard J, Isaksson OG, Rymo L. Regulation by GH of insulin-like growth factor-I mRNA expression in rat epiphyseal growth plate as studied with in situ hybridization. J. Endocrinol.125(1), 67–74 (1990).
- Yakar S, Liu JL, Stannard B et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl Acad. Sci. USA96(13), 7324–7329 (1999).
- Jenkins PJ, Mukherjee A, Shalet SM. Does growth hormone cause cancer? Clin. Endocrinol. (Oxf.)64(2), 115–121 (2006).
- Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr. Rev.28(1), 20–47 (2007).
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell100(1), 57–70 (2000).
- Harrela M, Koistinen H, Kaprio J et al. Genetic and environmental components of interindividual variation in circulating levels of IGF-I, IGF-II, IGFBP-1, and IGFBP-3. J. Clin. Invest.98(11), 2612–2615 (1996).
- Pollak M. The question of a link between insulin-like growth factor physiology and neoplasia. Growth Horm. IGF Res.10(Suppl. B), S21–S24 (2000).
- Laban C, Bustin SA, Jenkins PJ. The GH–IGF-I axis and breast cancer. Trends Endocrinol. Metab.14(1), 28–34 (2003).
- Yamauchi T, Ueki K, Tobe K et al. Tyrosine phosphorylation of the EGF receptor by the kinase Jak2 is induced by growth hormone. Nature390(6655), 91–96 (1997).
- Frank SJ. Mechanistic aspects of crosstalk between GH and PRL and ErbB receptor family signaling. J. Mammary Gland Biol. Neoplasia.13(1), 119–129 (2008).
- Brooks AJ, Wooh JW, Tunny KA, Waters MJ. Growth hormone receptor; mechanism of action. Int. J. Biochem. Cell Biol.40(10), 1984–1989 (2008).
- Gunnell D, Okasha M, Smith GD, Oliver SE, Sandhu J, Holly JM. Height, leg length, and cancer risk: a systematic review. Epidemiol. Rev.23(2), 313–342 (2001).
- Queiroga FL, Perez-Alenza D, Silvan G, Pena L, Lopes CS, Illera JC. Serum and intratumoural GH and IGF-I concentrations: Prognostic factors in the outcome of canine mammary cancer. Res. Vet. Sci.89(3), 396-403 (2010).
- Lundqvist E, Kaprio J, Verkasalo PK et al. Co-twin control and cohort analyses of body mass index and height in relation to breast, prostate, ovarian, corpus uteri, colon and rectal cancer among Swedish and Finnish twins. Int. J. Cancer121(4), 810–818 (2007).
- Key TJ, Appleby PN, Reeves GK, Roddam AW. Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: pooled individual data analysis of 17 prospective studies. Lancet Oncol.11(6), 530–542 (2010).
- Peyrat JP, Bonneterre J, Hecquet B et al. Plasma insulin-like growth factor-1 (IGF-1) concentrations in human breast cancer. Eur. J. Cancer29A(4), 492–497 (1993).
- Dupont J, Le Roith D. Insulin-like growth factor 1 and oestradiol promote cell proliferation of MCF-7 breast cancer cells: new insights into their synergistic effects. Mol. Pathol.54(3), 149–154 (2001).
- Byrne C, Colditz GA, Willett WC, Speizer FE, Pollak M, Hankinson SE. Plasma insulin-like growth factor (IGF) I, IGF-binding protein 3, and mammographic density. Cancer Res.60(14), 3744–3748 (2000).
- Kleinberg DL, Wood TL, Furth PA, Lee AV. Growth hormone and insulin-like growth factor-I in the transition from normal mammary development to preneoplastic mammary lesions. Endocr. Rev.30(1), 51–74 (2009).
- Rudd MF, Webb EL, Matakidou A et al. Variants in the GH–IGF axis confer susceptibility to lung cancer. Genome Res.16(6), 693–701 (2006).
- Le Marchand L, Donlon T, Seifried A, Kaaks R, Rinaldi S, Wilkens LR. Association of a common polymorphism in the human GH1 gene with colorectal neoplasia. J. Natl Cancer Inst.94(6), 454–460 (2002).
- Menashe I, Maeder D, Garcia-Closas M et al. Pathway analysis of breast cancer genome-wide association study highlights three pathways and one canonical signaling cascade. Cancer Res.70(11), 4453–4459 (2010).
- Canzian F, McKay JD, Cleveland RJ et al. Genetic variation in the growth hormone synthesis pathway in relation to circulating insulin-like growth factor-I, insulin-like growth factor binding protein-3, and breast cancer risk: results from the European prospective investigation into cancer and nutrition study. Cancer Epidemiol. Biomarkers Prev.14(10), 2316–2325 (2005).
- Diorio C, Brisson J, Berube S, Pollak M. Genetic polymorphisms involved in insulin-like growth factor (IGF) pathway in relation to mammographic breast density and IGF levels. Cancer Epidemiol. Biomarkers Prev.17(4), 880–888 (2008).
- Lai JH, Vesprini D, Zhang W, Yaffe MJ, Pollak M, Narod SA. A polymorphic locus in the promoter region of the IGFBP3 gene is related to mammographic breast density. Cancer Epidemiol. Biomarkers Prev.13(4), 573–582 (2004).
- Verheus M, Maskarinec G, Woolcott CG et al. IGF1, IGFBP1, and IGFBP3 genes and mammographic density: the Multiethnic Cohort. Int. J. Cancer127(5), 1115–1123 (2010).
- Morimoto LM, Newcomb PA, White E, Bigler J, Potter JD. Insulin-like growth factor polymorphisms and colorectal cancer risk. Cancer Epidemiol. Biomarkers Prev.14(5), 1204–1211 (2005).
- Friedrichsen DM, Hawley S, Shu J et al.IGF-I and IGFBP-3 polymorphisms and risk of prostate cancer. Prostate65(1), 44–51 (2005).
- Park K, Kim JH, Jeon HG, Byun SS, Lee E. Influence of IGFBP3 gene polymorphisms on IGFBP3 serum levels and the risk of prostate cancer in low-risk Korean men. Urology75(6), 1516, e1–e7 (2010).
- Rowlands MA, Gunnell D, Harris R, Vatten LJ, Holly JM, Martin RM. Circulating insulin-like growth factor peptides and prostate cancer risk: a systematic review and meta-analysis. Int. J. Cancer124(10), 2416–2429 (2009).
- Rinaldi S, Cleveland R, Norat T et al. Serum levels of IGF-I, IGFBP-3 and colorectal cancer risk: results from the EPIC cohort, plus a meta-analysis of prospective studies. Int. J. Cancer126(7), 1702–1715 (2010).
- Wolpin BM, Meyerhardt JA, Chan AT et al. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J. Clin. Oncol.27(2), 176–185 (2009).
- Stattin P, Bylund A, Rinaldi S et al. Plasma insulin-like growth factor-I, insulin-like growth factor-binding proteins, and prostate cancer risk: a prospective study. J. Natl Cancer Inst.92(23), 1910–1917 (2000).
- Renehan AG, Brennan BM. Acromegaly, growth hormone and cancer risk. Best Pract. Res. Clin. Endocrinol. Metab.22(4), 639–657 (2008).
- Loeper S, Ezzat S. Acromegaly: re-thinking the cancer risk. Rev. Endocr. Metab. Disord.9(1), 41–58 (2008).
- Colao A, Pivonello R, Auriemma RS et al. The association of fasting insulin concentrations and colonic neoplasms in acromegaly: a colonoscopy-based study in 210 patients. J. Clin. Endocrinol. Metab.92(10), 3854–3860 (2007).
- Shevah O, Laron Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: a preliminary report. Growth Horm. IGF Res.17(1), 54–57 (2007).
- Chernausek SD. Growth and development: how safe is growth hormone therapy for children? Nat. Rev. Endocrinol.6(5), 251–253 (2010).
- Bell J, Parker KL, Swinford RD, Hoffman AR, Maneatis T, Lippe B. Long-term safety of recombinant human growth hormone in children. J. Clin. Endocrinol. Metab.95(1), 167–177 (2010).
- Ergun-Longmire B, Mertens AC, Mitby P et al. Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J. Clin. Endocrinol. Metab.91(9), 3494–3498 (2006).
- Luft R, Olivecrona H. Hypophysectomy in the treatment of malignant tumors. Cancer10(4), 789–794 (1957).
- Moon HD, Simpson ME, Li CH, Evans HM. Neoplasms in rats treated with pituitary growth hormone; pulmonary and lymphatic tissues. Cancer Res.10(5), 297–308 (1950).
- Tornell J, Carlsson B, Pohjanen P, Wennbo H, Rymo L, Isaksson O. High frequency of mammary adenocarcinomas in metallothionein promoter-human growth hormone transgenic mice created from two different strains of mice. J. Steroid Biochem. Mol. Biol.43(1–3), 237–242 (1992).
- Bates P, Fisher R, Ward A, Richardson L, Hill DJ, Graham CF. Mammary cancer in transgenic mice expressing insulin-like growth factor II (IGF-II). Br. J. Cancer72(5), 1189–1193 (1995).
- Snibson KJ, Bhathal PS, Adams TE. Overexpressed growth hormone (GH) synergistically promotes carcinogen-initiated liver tumour growth by promoting cellular proliferation in emerging hepatocellular neoplasms in female and male GH-transgenic mice. Liver21(2), 149–158 (2001).
- Miquet JG, Gonzalez L, Matos MN et al. Transgenic mice overexpressing GH exhibit hepatic upregulation of GH-signaling mediators involved in cell proliferation. J. Endocrinol.198(2), 317–330 (2008).
- Hadsell DL, Murphy KL, Bonnette SG, Reece N, Laucirica R, Rosen JM. Cooperative interaction between mutant p53 and des(1–3)IGF-I accelerates mammary tumorigenesis. Oncogene19(7), 889–898 (2000).
- Carboni JM, Lee AV, Hadsell DL et al. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res.65(9), 3781–3787 (2005).
- Michaylira CZ, Simmons JG, Ramocki NM et al. Suppressor of cytokine signaling-2 limits intestinal growth and enterotrophic actions of IGF-I in vivo. Am. J. Physiol. Gastrointest. Liver Physiol.291(3), G472–G481 (2006).
- Ikeno Y, Hubbard GB, Lee S et al. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J. Gerontol. A Biol. Sci. Med. Sci.64(5), 522–529 (2009).
- Zhang X, Mehta RG, Lantvit DD et al. Inhibition of estrogen-independent mammary carcinogenesis by disruption of growth hormone signaling. Carcinogenesis28(1), 143–150 (2007).
- Yang XF, Beamer WG, Huynh H, Pollak M. Reduced growth of human breast cancer xenografts in hosts homozygous for the lit mutation. Cancer Res.56(7), 1509–1511 (1996).
- Thordarson G, Semaan S, Low C et al. Mammary tumorigenesis in growth hormone deficient spontaneous dwarf rats; effects of hormonal treatments. Breast Cancer Res. Treat.87(3), 277–290 (2004).
- Shen Q, Lantvit DD, Lin Q et al. Advanced rat mammary cancers are growth hormone dependent. Endocrinology148(10), 4536–4544 (2007).
- Wang Z, Prins GS, Coschigano KT et al. Disruption of growth hormone signaling retards early stages of prostate carcinogenesis in the C3(1)/T antigen mouse. Endocrinology146(12), 5188–5196 (2005).
- Pollak M, Blouin MJ, Zhang JC, Kopchick JJ. Reduced mammary gland carcinogenesis in transgenic mice expressing a growth hormone antagonist. Br. J. Cancer85(3), 428–430 (2001).
- Divisova J, Kuiatse I, Lazard Z et al. The growth hormone receptor antagonist pegvisomant blocks both mammary gland development and MCF-7 breast cancer xenograft growth. Breast Cancer Res. Treat.98(3), 315–327 (2006).
- McCutcheon IE, Flyvbjerg A, Hill H et al. Antitumor activity of the growth hormone receptor antagonist pegvisomant against human meningiomas in nude mice. J. Neurosurg.94(3), 487–492 (2001).
- Dagnaes-Hansen F, Duan H, Rasmussen LM, Friend KE, Flyvbjerg A. Growth hormone receptor antagonist administration inhibits growth of human colorectal carcinoma in nude mice. Anticancer Res.24(6), 3735–3742 (2004).
- de Ostrovich KK, Lambertz I, Colby JK et al. Paracrine overexpression of insulin-like growth factor-1 enhances mammary tumorigenesis in vivo. Am. J. Pathol.173(3), 824–834 (2008).
- Olivo-Marston SE, Hursting SD, Lavigne J et al. Genetic reduction of circulating insulin-like growth factor-1 inhibits azoxymethane-induced colon tumorigenesis in mice. Mol. Carcinog.48(12), 1071–1076 (2009).
- Wu Y, Yakar S, Zhao L, Hennighausen L, LeRoith D. Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis. Cancer Res.62(4), 1030–1035 (2002).
- Wu Y, Cui K, Miyoshi K et al. Reduced circulating insulin-like growth factor I levels delay the onset of chemically and genetically induced mammary tumors. Cancer Res.63(15), 4384–4388 (2003).
- Waters MJ. The growth hormone receptor. In: The Handbook of Physiology. Kostyo JL, Goodman HM (Eds). Oxford University Press, NY, USA, 397–444 (1999).
- Gebre-Medhin M, Kindblom LG, Wennbo H, Tornell J, Meis-Kindblom JM. Growth hormone receptor is expressed in human breast cancer. Am. J. Pathol.158(4), 1217–1222 (2001).
- Lin CJ, Mendonca BB, Lucon AM, Guazzelli IC, Nicolau W, Villares SM. Growth hormone receptor messenger ribonucleic acid in normal and pathologic human adrenocortical tissues – an analysis by quantitative polymerase chain reaction technique. J. Clin. Endocrinol. Metab.82(8), 2671–2676 (1997).
- Wu X, Liu F, Yao X, Li W, Chen C. Growth hormone receptor expression is up-regulated during tumorigenesis of human colorectal cancer. J. Surg. Res.143(2), 294–299 (2007).
- Harrison SM, Barnard R, Ho KY, Rajkovic I, Waters MJ. Control of growth hormone (GH) binding protein release from human hepatoma cells expressing full-length GH receptor. Endocrinology136(2), 651–659 (1995).
- Pazaitou-Panayiotou K, Kelesidis T, Kelesidis I et al. Growth hormone-binding protein is directly and IGFBP-3 is inversely associated with risk of female breast cancer. Eur. J. Endocrinol.156(2), 187–194 (2007).
- Lincoln DT, Sinowatz F, Kolle S, Takahashi H, Parsons P, Waters M. Up-regulation of growth hormone receptor immunoreactivity in human melanoma. Anticancer Res.19(3A), 1919–1931 (1999).
- Weiss-Messer E, Merom O, Adi A et al. Growth hormone (GH) receptors in prostate cancer: gene expression in human tissues and cell lines and characterization, GH signaling and androgen receptor regulation in LNCaP cells. Mol. Cell Endocrinol.220(1–2), 109–123 (2004).
- Mol JA, van Garderen E, Selman PJ, Wolfswinkel J, Rijinberk A, Rutteman GR. Growth hormone mRNA in mammary gland tumors of dogs and cats. J. Clin. Invest.95(5), 2028–2034 (1995).
- Mol JA, Henzen-Logmans SC, Hageman P, Misdorp W, Blankenstein MA, Rijnberk A. Expression of the gene encoding growth hormone in the human mammary gland. J. Clin. Endocrinol. Metab.80(10), 3094–3096 (1995).
- Queiroga FL, Perez-Alenza MD, Silvan G, Pena L, Lopes CS, Illera JC. Crosstalk between GH/IGF-I axis and steroid hormones (progesterone, 17β-estradiol) in canine mammary tumours. J. Steroid Biochem. Mol. Biol.110(1–2), 76–82 (2008).
- Raccurt M, Lobie PE, Moudilou E et al. High stromal and epithelial human GH gene expression is associated with proliferative disorders of the mammary gland. J. Endocrinol.175(2), 307–318 (2002).
- Ratkaj I, Stajduhar E, Vucinic S et al. Integrated gene networks in breast cancer development. Funct. Integr. Genomics10(1), 11–19 (2010).
- van Garderen E, Schalken JA. Morphogenic and tumorigenic potentials of the mammary growth hormone/growth hormone receptor system. Mol. Cell Endocrinol.197(1–2), 153–165 (2002).
- Gil-Puig C, Blanco M, Garcia-Caballero T, Segura C, Perez-Fernandez R. Pit-1/GHF-1 and GH expression in the MCF-7 human breast adenocarcinoma cell line. J. Endocrinol.173(1), 161–167 (2002).
- Chopin LK, Veveris-Lowe TL, Philipps AF, Herington AC. Co-expression of GH and GHR isoforms in prostate cancer cell lines. Growth Horm. IGF Res.12(2), 126–136 (2002).
- Colao A, Marzullo P, Spiezia S et al. Effect of two years of growth hormone and insulin-like growth factor-I suppression on prostate diseases in acromegalic patients. J. Clin. Endocrinol. Metab.85(10), 3754–3761 (2000).
- Colao A, Di Somma C, Spiezia S, Filippella M, Pivonello R, Lombardi G. Effect of growth hormone (GH) and/or testosterone replacement on the prostate in GH-deficient adult patients. J. Clin. Endocrinol. Metab.88(1), 88–94 (2003).
- Wang W, Edington HD, Rao UN et al. Effects of high-dose IFNα2β on regional lymph node metastases of human melanoma: modulation of STAT5, FOXP3, and IL-17. Clin. Cancer Res.14(24), 8314–8320 (2008).
- Ruan W, Powell-Braxton L, Kopchick JJ, Kleinberg DL. Evidence that insulin-like growth factor I and growth hormone are required for prostate gland development. Endocrinology140(5), 1984–1989 (1999).
- Cunningham BC, Bass S, Fuh G, Wells JA. Zinc mediation of the binding of human growth hormone to the human prolactin receptor. Science250(4988), 1709–1712 (1990).
- Zhu T, Starling-Emerald B, Zhang X et al. Oncogenic transformation of human mammary epithelial cells by autocrine human growth hormone. Cancer Res.65(1), 317–324 (2005).
- Xu XQ, Emerald BS, Goh EL et al. Gene expression profiling to identify oncogenic determinants of autocrine human growth hormone in human mammary carcinoma. J. Biol. Chem.280(25), 23987–24003 (2005).
- Perry JK, Mohankumar KM, Emerald BS, Mertani HC, Lobie PE. The contribution of growth hormone to mammary neoplasia. J. Mammary Gland Biol. Neoplasia.13(1), 131–145 (2008).
- Brunet-Dunand SE, Vouyovitch C, Araneda S et al. Autocrine human growth hormone promotes tumor angiogenesis in mammary carcinoma. Endocrinology150(3), 1341–1352 (2009).
- Kaulsay KK, Zhu T, Bennett W, Lee KO, Lobie PE. The effects of autocrine human growth hormone (hGH) on human mammary carcinoma cell behavior are mediated via the hGH receptor. Endocrinology142(2), 767–777 (2001).
- van den Eijnden MJ, Strous GJ. Autocrine growth hormone: effects on growth hormone receptor trafficking and signaling. Mol. Endocrinol.21(11), 2832–2846 (2007).
- Conway-Campbell BL, Wooh JW, Brooks AJ et al. Nuclear targeting of the growth hormone receptor results in dysregulation of cell proliferation and tumorigenesis. Proc. Natl Acad. Sci. USA104(33), 13331–13336 (2007).
- Mertani HC, Garcia-Caballero T, Lambert A et al. Cellular expression of growth hormone and prolactin receptors in human breast disorders. Int. J. Cancer79(2), 202–211 (1998).
- Lincoln DT, Kaiser HE, Raju GP, Waters MJ. Growth hormone and colorectal carcinoma: localization of receptors. In Vivo14(1), 41–49 (2000).
- Garcia-Caballero T, Mertani HM, Lambert A et al. Increased expression of growth hormone and prolactin receptors in hepatocellular carcinomas. Endocrine12(3), 265–271 (2000).
- Dehari R, Nakamura Y, Okamoto N, Nakayama H. Increased nuclear expression of growth hormone receptor in uterine cervical neoplasms of women under 40 years old. Tohoku J. Exp. Med.216(2), 165–172 (2008).
- Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer94(2), 184–188 (2006).
- Conway-Campbell BL, Brooks AJ, Robinson PJ, Perani M, Waters MJ. The extracellular domain of the growth hormone receptor interacts with coactivator activator to promote cell proliferation. Mol. Endocrinol.22(9), 2190–2202 (2008).
- Sui Y, Yang Z, Xiong S et al. Gene amplification and associated loss of 5´ regulatory sequences of CoAA in human cancers. Oncogene26(6), 822–835 (2007).
- Li X, Hoeppner LH, Jensen ED, Gopalakrishnan R, Westendorf JJ. Co-activator activator (CoAA) prevents the transcriptional activity of Runt domain transcription factors. J. Cell. Biochem.108(2), 378–387 (2009).
- Auboeuf D, Dowhan DH, Li X et al. CoAA, a nuclear receptor coactivator protein at the interface of transcriptional coactivation and RNA splicing. Mol. Cell Biol.24(1), 442–453 (2004).
- Carver KC, Schuler LA. Prolactin does not require insulin-like growth factor intermediates but synergizes with insulin-like growth factor I in human breast cancer cells. Mol. Cancer Res.6(4), 634–643 (2008).
- Carver KC, Arendt LM, Schuler LA. Complex prolactin crosstalk in breast cancer: new therapeutic implications. Mol. Cell Endocrinol.307(1–2), 1–7 (2009).
- Tornell J, Rymo L, Isaksson OG. Induction of mammary adenocarcinomas in metallothionein promoter-human growth hormone transgenic mice. Int. J. Cancer49(1), 114–117 (1991).
- Wennbo H, Gebre-Medhin M, Gritli-Linde A, Ohlsson C, Isaksson OG, Tornell J. Activation of the prolactin receptor but not the growth hormone receptor is important for induction of mammary tumors in transgenic mice. J. Clin. Invest.100(11), 2744–2751 (1997).
- Clevenger CV. Role of prolactin/prolactin receptor signaling in human breast cancer. Breast Dis.18, 75–86 (2003).
- Dagvadorj A, Collins S, Jomain JB et al. Autocrine prolactin promotes prostate cancer cell growth via Janus kinase-2-signal transducer and activator of transcription-5a/b signaling pathway. Endocrinology148(7), 3089–3101 (2007).
- Rouet V, Bogorad RL, Kayser C et al. Local prolactin is a target to prevent expansion of basal/stem cells in prostate tumors. Proc. Natl Acad. Sci. USA107(34), 15199–15204 (2010).
- Harbaum L, Pollheimer MJ, Bauernhofer T et al. Clinicopathological significance of prolactin receptor expression in colorectal carcinoma and corresponding metastases. Mod. Pathol.23(7), 961–971 (2010).
- Mohankumar KM, Perry JK, Kannan N et al. Transcriptional activation of signal transducer and activator of transcription (STAT) 3 and STAT5B partially mediate homeobox A1-stimulated oncogenic transformation of the immortalized human mammary epithelial cell. Endocrinology149(5), 2219–2229 (2008).
- Tang JZ, Zuo ZH, Kong XJ et al. Signal transducer and activator of transcription (STAT)-5A and STAT5B differentially regulate human mammary carcinoma cell behavior. Endocrinology151(1), 43–55 (2010).
- Tang JZ, Kong XJ, Banerjee A et al. STAT3a is oncogenic for endometrial carcinoma cells and mediates the oncogenic effects of autocrine human growth hormone. Endocrinology151(9), 4133–4145 (2010).
- Levine RL, Gilliland DG. JAK-2 mutations and their relevance to myeloproliferative disease. Curr. Opin Hematol.14(1), 43–47 (2007).
- Morgan KJ, Gilliland DG. A role for JAK2 mutations in myeloproliferative diseases. Annu. Rev. Med.59, 213–222 (2008).
- Lacronique V, Boureux A, Valle VD et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science278(5341), 1309–1312 (1997).
- Carlesso N, Frank DA, Griffin JD. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J. Exp. Med.183(3), 811–820 (1996).
- Pikman Y, Lee BH, Mercher T et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med.3(7), e270 (2006).
- Funakoshi-Tago M, Tago K, Abe M, Sonoda Y, Kasahara T. STAT5 activation is critical for the transformation mediated by myeloproliferative disorder-associated JAK2 V617F mutant. J. Biol. Chem.285(8), 5296–5307 (2010).
- Hoelbl A, Schuster C, Kovacic B et al. STAT5 is indispensable for the maintenance of Bcr/Abl-positive leukaemia. EMBO Mol. Med.2(3), 98–110 (2010).
- Schwaller J, Parganas E, Wang D et al. STAT5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Mol. Cell6(3), 693–704 (2000).
- Gu L, Vogiatzi P, Puhr M et al. STAT5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocr. Relat. Cancer17(2), 481–493 (2010).
- Mirmohammadsadegh A, Hassan M, Bardenheuer W et al. STAT5 phosphorylation in malignant melanoma is important for survival and is mediated through Srs and Jak1 kinases. J. Invest. Dermatol.126(10), 2272–2280 (2006).
- Hassel JC, Winnemoller D, Schartl M, Wellbrock C. STAT5 contributes to antiapoptosis in melanoma. Melanoma Res.18(6), 378–385 (2008).
- Lee TK, Man K, Poon RT et al. Signal transducers and activators of transcription 5b activation enhances hepatocellular carcinoma aggressiveness through induction of epithelial-mesenchymal transition. Cancer Res.66(20), 9948–9956 (2006).
- Wagner KU, Rui H. Jak2/STAT5 signaling in mammogenesis, breast cancer initiation and progression. J. Mammary Gland Biol. Neoplasia.13(1), 93–103 (2008).
- Koppikar P, Lui VW, Man D et al. Constitutive activation of signal transducer and activator of transcription 5 contributes to tumor growth, epithelial–mesenchymal transition, and resistance to epidermal growth factor receptor targeting. Clin. Cancer Res.14(23), 7682–7690 (2008).
- Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr. Opin Cell Biol.20(2), 150–155 (2008).
- Mallette FA, Gaumont-Leclerc MF, Huot G, Ferbeyre G. Myc down-regulation as a mechanism to activate the Rb pathway in STAT5a-induced senescence. J. Biol. Chem.282(48), 34938–34944 (2007).
- Mallette FA, Moiseeva O, Calabrese V, Mao B, Gaumont-Leclerc MF, Ferbeyre G. Transcriptome analysis and tumor suppressor requirements of STAT5-induced senescence. Ann. NY Acad. Sci.1197, 142–151 (2010).
- Schartl M, Wilde B, Laisney JA, Taniguchi Y, Takeda S, Meierjohann S. A mutated EGFR is sufficient to induce malignant melanoma with genetic background-dependent histopathologies. J. Invest. Dermatol.130(1), 249–258 (2010).
- Mohapatra S, Coppola D, Riker AI, Pledger WJ. Roscovitine inhibits differentiation and invasion in a three-dimensional skin reconstruction model of metastatic melanoma. Mol. Cancer Res.5(2), 145–151 (2007).
- Senderowicz AM. Small-molecule cyclin-dependent kinase modulators. Oncogene22(42), 6609–6620 (2003).
- Mohapatra S, Chu B, Wei S et al. Roscovitine inhibits STAT5 activity and induces apoptosis in the human leukemia virus type 1-transformed cell line MT-2. Cancer Res.63(23), 8523–8530 (2003).
- Ahonen TJ, Xie J, LeBaron MJ et al. Inhibition of transcription factor STAT5 induces cell death of human prostate cancer cells. J. Biol. Chem.278(29), 27287–27292 (2003).
- Dagvadorj A, Kirken RA, Leiby B, Karras J, Nevalainen MT. Transcription factor signal transducer and activator of transcription 5 promotes growth of human prostate cancer cells in vivo. Clin. Cancer Res.14(5), 1317–1324 (2008).
- Kazansky AV, Spencer DM, Greenberg NM. Activation of signal transducer and activator of transcription 5 is required for progression of autochthonous prostate cancer: evidence from the transgenic adenocarcinoma of the mouse prostate system. Cancer Res.63(24), 8757–8762 (2003).
- Ginestier C, Wicha MS. Mammary stem cell number as a determinate of breast cancer risk. Breast Cancer Res.9(4), 109 (2007).
- Tan SH, Dagvadorj A, Shen F et al. Transcription factor STAT5 synergizes with androgen receptor in prostate cancer cells. Cancer Res.68(1), 236–248 (2008).
- Yamashita H, Iwase H, Toyama T, Fujii Y. Naturally occurring dominant-negative STAT5 suppresses transcriptional activity of estrogen receptors and induces apoptosis in T47D breast cancer cells. Oncogene22(11), 1638–1652 (2003).
- Iavnilovitch E, Groner B, Barash I. Overexpression and forced activation of stat5 in mammary gland of transgenic mice promotes cellular proliferation, enhances differentiation, and delays postlactational apoptosis. Mol. Cancer Res.1(1), 32–47 (2002).
- Iavnilovitch E, Cardiff RD, Groner B, Barash I. Deregulation of STAT5 expression and activation causes mammary tumors in transgenic mice. Int. J. Cancer112(4), 607–619 (2004).
- Pardanani AD, Levine RL, Lasho T et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood108(10), 3472–3476 (2006).
- Ding J, Komatsu H, Wakita A et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood103(11), 4198–4200 (2004).
- Bernichtein S, Touraine P, Goffin V. New concepts in prolactin biology. J. Endocrinol.206(1), 1–11 (2010).
- Lempereur L, Brambilla D, Scoto GM et al. Growth hormone protects human lymphocytes from irradiation-induced cell death. Br. J. Pharmacol.138(8), 1411–1416 (2003).
- Wu X, Wan M, Li G et al. Growth hormone receptor overexpression predicts response of rectal cancers to pre-operative radiotherapy. Eur. J. Cancer42(7), 888–894 (2006).
- Zatelli MC, Minoia M, Mole D et al. Growth hormone excess promotes breast cancer chemoresistance. J. Clin. Endocrinol. Metab.94(10), 3931–3938 (2009).
- Blackmore DG, Golmohammadi MG, Large B, Waters MJ, Rietze RL. Exercise increases neural stem cell number in a growth hormone-dependent manner, augmenting the regenerative response in aged mice. Stem Cells27(8), 2044–2052 (2009).
Website
- Oncomine database www.oncomine.org