Abstract
The diagnosis and treatment of medullary thyroid cancer (MTC) has changed considerably over the past 40 years, although survival has not improved over the past three decades. From the first clinical descriptions of MTC and the multiple endocrine neoplasia syndromes to the identification of the RET proto-oncogene, the concept of prophylactic thyroidectomy has become a reality for some families. The surgery is usually performed by high-volume surgeons in specialized centers with results that rarely leave patients with complications but with maximum survival benefit. For patients with unresectable or metastatic disease, many new targeted agents have been developed, some of which have shown great promise and are close to being approved for a disease which hitherto has had no systemic treatment options. This article highlights the important advances in the diagnosis and management of MTC.
Medscape: Continuing Medical Education Online
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Expert Reviews Ltd. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 70% minimum passing score and complete the evaluation at www.medscape.org/journal/expertendo; (4) view/print certificate.
Release date: 7 July 2011; Expiration date: 7 July 2012
Learning objectives
Upon completion of this activity, participants will be able to:
• Analyze the epidemiology and prognosis of MTC
• Apply current recommendations regarding prophylactic thyroidectomy for patients at high risk for MTC
• Distinguish means to diagnose MTC
• Evaluate treatment options for MTC
Financial & competing interests disclosure
EDITOR
Elisa Manzotti
Editorial Director, Future Science Group, London, UK.
Disclosure: Elisa Manzotti has disclosed no relevant financial relationships.
CME AUTHOR
Charles P Vega, MD
Associate Professor; Residency Director, Department of Family Medicine, University of California, Irvine, CA, USA.
Disclosure: Charles P Vega has disclosed no relevant financial relationships.
AUTHORS AND CREDENTIALS
Hari A Deshpande, MD
Smilow Cancer Hospital, Section of Endocrine Oncology, Yale School of Medicine, Yale University, Departments of Medical Oncology and Surgery, 333 Cedar Street, FMP 124, New Haven, CT 06520, USA.
Disclosure: Hari A Deshpande has disclosed no relevant financial relationships.
Daniel Morgensztern, MD
Smilow Cancer Hospital, Section of Endocrine Oncology, Yale School of Medicine, Yale University, Departments of Medical Oncology and Surgery, 333 Cedar Street, FMP 121, New Haven, CT 06520, USA.
Disclosure: Daniel Morgensztern has disclosed no relevant financial relationships.
Julie Ann Sosa, MD
Departments of Surgery and Medical Oncology, Yale School of Medicine and Smilow Cancer Hospital at Yale-New Haven, 333 Cedar Street, TMP 204, New Haven, CT 06520, USA.
Disclosure: Julie Ann Sosa has disclosed no relevant financial relationships.
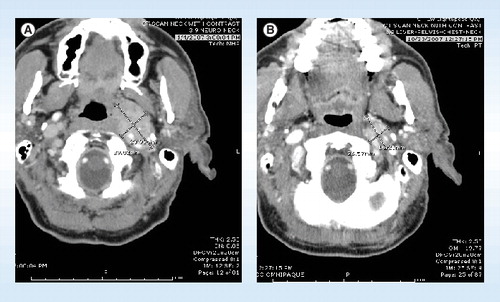
(A) Shows the patient pre-treatment and (B) post-treatment, with significant shrinkage in the size of the tumor.
Characterization of medullary thyroid cancer
Medullary thyroid cancer (MTC) accounts for less than 10% of all incident cases of thyroid malignancy but 13.4% of thyroid cancer-related deaths Citation[1]. Indeed, with the rapid increase in incidence of papillary thyroid cancer (PTC), the relative proportion of new cases of thyroid cancer that are MTC has fallen to approximately 3% Citation[2]. This indolent malignancy differs from other types of thyroid cancer in that it originates from the parafollicular C cells that produce calcitonin and not iodine-rich thyroid hormone, making it not susceptible to treatment with radioactive iodine. The pathogenesis of this disease has been extensively documented over the past 70 years. Soon after the earliest descriptions of this disease in 1959 Citation[3], there were reports of a familial syndrome, initially in a single family Citation[4] and subsequently associated with other disorders, including phaeochromocytomas and primary hyperparathyroidism Citation[5,6]. It is now clear that there are multiple hereditary forms of this cancer, including isolated familial MTC (FMTC) and the multiple endocrine neoplasia syndromes (MEN 2A and MEN 2B), although the majority of patients (∼75%) have sporadic MTC Citation[7] . MEN 2A accounts for 60% of the hereditary forms, MEN 2B 5% and FMTC 35% Citation[8].
Distant metastatic disease is the main cause of death in patients with MTC, occurring in approximately half of incident cases at presentation and often involving multiple organs such as the liver, lungs and bones Citation[9]. Extra-cervical metastases and unresectable advanced local disease are essentially incurable. Consequently, the best chance of curing patients with the disease is to identify patients at an early stage when the tumor is amenable to resection, especially if it is confined to the thyroid. In a review of over 1200 cases, the mean survival time after the diagnosis of MTC was 8.6 years (range, 0–29.6 years). Patients with tumors confined to the thyroid gland had a 10-year survival rate of 95.6%, whereas patients with regional stage disease had an overall survival rate of 75.5%. Patients with distant metastases at diagnosis had a poor prognosis, with only 40% surviving for 10 years Citation[10]. Although traditional cytotoxic chemotherapy and radiation techniques have been historically ineffective in MTC, novel small-molecule tyrosine kinase inhibitors appear to hold promise Citation[11].
Discovery of RET
Until recently, it was likely that the detection of tumor at an early stage occurred either by the incidental finding of a thyroid nodule or through screening of family members of an index patient who had previously been diagnosed with MTC or with the MEN 2 syndrome. Previous work had shown that the MEN syndromes are inherited in an autosomal dominant manner Citation[12], suggesting that 50% of patients would be screened unnecessarily. The yield from screening was improved with the discovery in 1985 of a new human transforming gene detected by transfection of NIH 3T3 cells with a lymphoma DNA Citation[13]. Subsequent work pinpointed mutations on chromosome 10, followed by the identification of germline mutations in the RET (rearranged during transfection) proto-oncogene, located at 10q11.2, in patients with MEN 2A, MEN 2B and FMTC Citation[6].
Prophylactic thyroidectomy
This historic chain of events allowed the accurate screening of patients and families to document the mutation in the index case and the subsequent identification of those who would be at risk of developing the disease. Prophylactic thyroidectomy could therefore potentially prevent this disease from occurring in family members of patients with MTC. Since prophylactic thyroidectomy often involves surgery on children under the age of 5 years, the American Thyroid Association (ATA) has published guidelines to determine both the indications and timing for the surgery Citation[14]. Potential complications following thyroidectomy include temporary or permanent recurrent laryngeal nerve injury resulting in dysphonia and hypoparathyroidism, which can be especially difficult to manage in children and may interfere with quality of life for decades. In addition, these patients require lifelong thyroid hormone replacement. Moreover, some RET codon mutations are associated with earlier onset of a more aggressive disease; this is especially so for codon 918 mutations.
The ATA guidelines stratify patients according to the mutation found and consider allowing a delay in thyroidectomy for lower risk mutations in selected patients, including those with a normal annual serum calcitonin in addition to a normal annual neck ultrasound, a less aggressive family history and family preference Citation[13]. Mutations are classified based on time to development of MTC and are stratified from A (lowest risk) to D (highest risk). Risk D mutations require prophylactic thyroidectomy at detection or within the first year of life, while the ATA suggests surgery before the age of 5 years for A, B and C mutations, with specific clinical criteria for exceptions Citation[13]. In patients with hereditary or sporadic MTC diagnosed clinically, 80% have already metastasized to ipsilateral and 40% to contralateral cervical lymph nodes Citation[15]. In these cases, the ATA guidelines suggest therapeutic total thyroidectomy and central neck dissection, along with lateral compartment lymph node dissection for clinically or radiographically evident lymphadenopathy. Subsequent studies have shown that if all these criteria are met, the guidelines offer reasonable mutation-based recommendations to guide timing of prophylactic thyroidectomy. Safe delay of thyroidectomy beyond the age of 5 years is possible if all postponement guidelines are met, particularly for lower risk level A and B mutations. Additional data are needed to determine whether individuals with level C mutations who are 5 years of age and older can be safely monitored Citation[16].
Surgeons with a variety of training backgrounds, including pediatric surgeons, otolaryngologists head and neck surgeons, general surgeons and endocrine surgeons perform thyroidectomies in pediatric patients. Some surgeon characteristics appear to be associated with improved patient outcomes. A multidisciplinary approach that involves pediatric endocrinologists, pediatricians, surgeons, nuclear medicine physicians, anesthesiologists and pathologists is an essential part of the preoperative, postoperative and long-term management of children with thyroid disease, and this is especially important for those pediatric patients who undergo thyroidectomy Citation[17].
As in adults, surgeon volume appears to be independently associated in a dose-response fashion with patient outcomes in children undergoing thyroidectomy Citation[18]. In a study using national data with patients and surgeons from all types of healthcare settings, surgeon volume (in adults and children) had a greater impact on patient outcomes than surgeon specialty Citation[19]. In-hospital endocrine-related complication rates tended to be lower for high-volume surgeons compared with low-volume surgeons (5.6 vs 11%; p = not significant). Of note, these complication rates are higher than the rates of 1–2% reported for adults undergoing similar procedures Citation[20]. High-volume surgeons had significantly shorter length of hospital stay (1.5 vs 2.1 days; p < 0.05) and costs (US$12,474 vs 15,662; p < 0.05) than low-volume surgeons. The challenge in pediatric endocrine surgery is that overall experience is limited because pediatric endocrine diseases requiring operative intervention are relatively rare. These findings suggest that surgeons with the best outcomes should be selected for thyroid operations on children and adults.
In a literature review examining over 20 case series reports on a total of 1800 pediatric patients, rates of permanent recurrent laryngeal nerve injury and permanent hypoparathyroidism were lowest in operations by high-volume thyroid surgeons Citation[21]. The reported range of complication rates was considerable: nerve injury rates ranged from 0 to 40%, and hypoparathyroidism rates varied from 0 to 32%. The authors go on to note that “all surgery should be performed by high-volume, experienced endocrine, pediatric and head and neck surgeons.” In a review article supporting thyroidectomy as the optimal treatment for pediatric Graves’ disease, Lee et al. suggested that patients travel outside their local area if necessary to find surgeons with extensive experience in pediatric thyroidectomy; the same conclusion would certainly apply to patients with MTC Citation[22].
Published guidelines, including the ATA management guidelines for patients with thyroid nodules and differentiated thyroid cancer and the National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology, are clear in acknowledging the importance of surgeon volume for patient outcomes in adults undergoing thyroidectomy Citation[23,24]. Fewer studies have examined the effect of surgeon volume on surgical outcomes specifically among pediatric patients. The ATA Medullary Thyroid Cancer Management Guidelines address the role of surgeon experience in pediatric outcomes, asserting that “children undergoing thyroidectomy … have higher complication rates than adults and have better outcomes when operated on by high-volume surgeons Citation[13].” Surgeon volume appears to be the most important and robust predictor of pediatric outcomes after thyroidectomy and it appears to be independent of the effect of surgeon specialty.
Calcitonin as a screening test
Multiple European studies have shown that the routine measurement of serum calcitonin in patients with thyroid nodules is effective in the detection of clinically occult MTC Citation[25–31]. Further studies have demonstrated that this was associated with an improvement in patient outcome Citation[32]. In 2006, the European Thyroid Association and Cancer Research Network followed the lead of individual European countries and developed a European consensus report on the management of differentiated thyroid carcinoma, including the use of calcitonin screening in patients with thyroid nodules. These guidelines recommended the use of calcitonin measurement in the initial diagnostic evaluation of thyroid nodules Citation[33]. In the USA, the ATA management guidelines for patients with thyroid nodules could not “recommend either for or against the routine measurement of serum calcitonin” due to unresolved issues of sensitivity, specificity, assay performance, cost–effectiveness and lack of pentagastrin availability in the USA. Calcium infusion (using, for example, the Doyle protocol) has been used as an alternative.
There are many difficulties associated with using calcitonin as a screening tool, in part related to its lack of perfect specificity or sensitivity with regard to MTC. As a result, many thyroidologists favor screening with a combination of basal and stimulated calcitonin levels. For example, nonhereditary C-cell hyperplasia has been associated with PTC and lymphocytic (Hashimoto’s) thyroiditis. In addition, serum calcitonin can be elevated in a variety of systemic conditions, including chronic renal insufficiency, patients taking proton pump inhibitors with hypergastrinemia, critical illness, smoking, and in patients with neuroendocrine tumors of the lung, pancreas and prostate. Most centers agree that a basal calcitonin value of greater than 100 pg/ml is highly suggestive of MTC; the challenge is interpretation of values between 20 and 100 pg/ml. It is also important to note that while a large portion of the (older) literature is based on the results of calcitonin two-site radioimmunometric assays, most contemporary testing in the USA and much of Europe is based on an automated two-site chemiluminescent assay; this complicates further comparison of data.
A recent cost–effectiveness study demonstrated that the addition of calcitonin screening to current ATA guidelines for the evaluation of thyroid nodules would cost US$11,793 per life-year-saved (LYS) (range: US$10,941–12,646); when extrapolated to the national level, calcitonin screening for MTC would yield an additional 113,000 life-years at a cost increase of 5.3% Citation[34]. The cost–effectiveness of screening programs for breast and colon cancer ranges from US$3400–28,700 to US$10,000–25,000 per LYS, respectively Citation[35–37]. Screening for mild hypothyroidism using serum thyroid-stimulating hormone measurement has a cost–effectiveness of US$9223–20,000 per LYS. Calcitonin screening is sensitive to patient age, gender and changes in disease prevalence, specificity of fine-needle aspiration (FNA) and calcitonin screening, serum calcitonin screening level, test costs and length of follow-up Citation[24].
Diagnosis of MTC
The diagnosis of MTC should be anticipated first based on a suggestive family history, especially when combined with a physical exam that includes a palpable anterior neck mass corresponding to a thyroid nodule with or without associated lymphadenopathy in the supraclavicular or cervical chains. Symptoms of flushing and diarrhea should also raise concern for a diagnosis of MTC. Indeed, the symptoms are not due to hypercalcitoninemia but rather to the production of other enteric hormones. Ultrasound in the hands of an experienced radiologist, endocrinologist or surgeon can be helpful, although two recent studies suggested that there is significant overlap between the ultrasonographic characteristics of PTC and MTC Citation[38,39]. Nodules containing MTC may be more ovoid or ‘more tall than wide,’ and they may also be larger and more often cystic than those with PTC. Coarse calcifications may also be more prevalent in MTC.
Ultrasound should be combined with FNA, although there is controversy regarding the sensitivity of FNA for MTC. In truth, the diagnostic accuracy of the technique likely varies in clinical practice. Combined data for the three largest cytopathology series with surgical follow-up indicate that 79% of patients had FNAs that were positive for MTC and 96% suggested some form of thyroid lesion (i.e., an ‘indeterminate’ nodule) that prompted surgical intervention for diagnosis and/or treatment Citation[2,40–42]. Routine cytological specimens are not usually stained for calcitonin unless there is a suspicion of MTC, although measurement of calcitonin in the fluid aspirate has been demonstrated as a diagnostic tool for the diagnosis of MTC in a thyroid nodule Citation[2,43–47].
Evaluation once the diagnosis of MTC is made
Once the diagnosis of MTC is made, ATA guidelines recommend the measurement of the serum tumor markers calcitonin and carcinoembryonic antigen (CEA). These tumor markers are useful for the purposes of prognosis, prediction of the likelihood of metastatic disease and for comparison with postoperative levels in order to measure the adequacy of resection. RET proto-oncogene testing is also recommended in all of these patients.
As noted previously, a positive RET test result would provide critical risk information regarding concurrent pheochromocytoma and hyperparathyroidism; if a pheochromocytoma were to be identified, adrenalectomy following appropriate medical preparation (α with/without β-blockade, as well as hydration) should be performed prior to thyroidectomy; parathyroidectomy could be performed concurrently with thyroid resection. RET results would also provide valuable information about potential bilobar involvement in the thyroid and disease risk in first-degree relatives, in which case genetic counseling should be offered and informed screening performed. RET testing is also important on tumoral tissue to determine the presence of somatic mutations that may be used to guide treatment options. In MTC patients with a negative family history for the disease, the estimated risk of familial involvement is 5–6% Citation[48]. All patients with MTC should therefore be tested for a de novo RET mutation.
Definitive surgery for newly diagnosed MTC
A difference in opinion remains as to the extent of surgery needed for patients with MTC. Most surgeons recommend a minimum of total thyroidectomy with a central neck dissection for patients with MTC larger than 1.5 cm. The central neck compartment (level VI lymph nodes) is an area of particular interest in endocrine surgery, largely owing to debate about its appropriate role for PTC; as a result, its landmarks have been defined by consensus. Certainly, those patients with inherited forms of MTC are at a higher risk for multifocal disease arising from a background of C-cell hyperplasia, enhancing the importance of performing a total thyroidectomy. The value of a lateral neck dissection (i.e., modified radical neck dissection for compartmental evacuation of lymph nodes in levels II, III, IV and V; rarely is level I involved in thyroid cancer) is often determined on a case-by-case basis, based on whether there is clinical or radiographic evidence for pathologic lymphadenopathy and also based on the absolute calcitonin level Citation[14].
However, one of the concerns with this approach is that an incomplete resection can compromise the chance of curing the patient of the disease. The benefits of additional surgery need to be weighed carefully against the attendant risks of recurrent laryngeal nerve injury and hypoparathyroidism during central neck dissection and injury to the spinal accessory nerve, brachial plexus, phrenic nerve, carotid sheath and thoracic duct (on the left) during lateral neck exploration. Some groups have advocated a more aggressive surgical approach up front, based on lower calcitonin thresholds. For example, in one of the largest single institution series of 300 consecutive patients with previously untreated MTC, Machens and Dralle demonstrated that most newly diagnosed MTC patients with pretherapeutic basal calcitonin levels of at least 200 pg/ml may need bilateral compartment-oriented neck surgery to reduce the number of reoperations. Lymph node metastases were present in the bilateral central and lateral neck, contralateral central neck and contralateral lateral neck and upper mediastinum, beyond basal calcitonin thresholds of 20, 50, 200 and 500 pg/ml, respectively Citation[49].
Once definitive surgery has been performed, patients can be monitored with regular history and physical examinations at office visits, as well as checking calcitonin and CEA levels. Calcitonin levels decline progressively and variably after surgery when all or most MTC has been resected, with some patients not reaching their nadir after surgery for up to 8–12 weeks or longer. Repeat calcitonin and CEA values should be obtained again at that point, as they are useful going forward for calculating calcitonin doubling times after approximately four measurements. Based on the ATA consensus practice guidelines, if the calcitonin value exceeds 400 pg/ml, patients should be considered for additional imaging, including neck computed tomography (CT) or MRI, chest CT, liver protocol abdominal MRI and bone imaging.
If these levels are elevated after surgery, they should be followed every 6–12 months, although additional more frequent studies may be needed for rising values. If the levels are stable, no further imaging is required. Patients with undetectable levels following surgery can have their calcitonin and CEA tests monitored annually. Whenever serum calcitonin becomes undetectable (either basal or, even better, after pentagastrin stimulation), the patient is probably cured and has no more than a 5% chance of recurrence Citation[50]. Calcitonin doubling times of greater than 2 years seem to be associated with a better long-term prognosis than those less than 6 months. These tumor marker levels, in turn, can be employed to inform whether additional imaging such as cervical ultrasound and/or CT of the neck, chest and abdomen (with or without liver MRI) is necessary, based on the level of suspicion for persistent or recurrent disease.
In approximately 10–15% of cases, the diagnosis of MTC is only made after thyroidectomy. This is higher than the 0.14% prevalence of occult MTC recently noted among unselected autopsy specimens from 24 autopsy series Citation[51]. As Valle and Kloos note, their observed prevalence may be an underestimate, owing to the variability between the series in terms of slice thickness, calcitonin immunostaining and extent of the pathological exam of the thyroid Citation[51]. The delay in diagnosis among patients whose MTC is not diagnosed until the return of the final surgical pathology can have adverse consequences, as it can result in inadequate surgery at the primary operation, necessitating remedial surgery and the consequent elevation in risk of hypoparathyrodism and nerve injury. In addition, there is the potentially life-threatening risk of missing a coexistent pheochromocytoma in those patients who might have unrecognized MEN 2 syndrome and primary hyperparathyroidism in patients who have MEN 2A syndrome.
Systemic therapy for advanced disease
Traditional chemotherapy
Chemotherapy agents, including doxorubicin and cisplatin, have been evaluated in the systemic treatment of MTC. While the medullary subgroup of thyroid cancers was thought to have the best response rates compared with differentiated and anaplastic types, no definitive studies have demonstrated a long-term benefit. It has been suggested that responders to chemotherapy can see an improvement in overall survival from 5–15 months, although randomized studies proving this have been lacking Citation[52]. Other agents such as paclitaxel and gemcitabine that have broad activity in many cancers have been disappointing with regard to their effect on MTC Citation[53].
The authors have seen individual patients respond to oral chemotherapy agents, as measured by biochemical and radiological criteria [Deshpande HA, Morgensztern D, Sosa JA, Unpublished Data]; this is consistent with published reports showing progression-free intervals of 11 months–4 years in patients treated with the thymidylate synthetase inhibitor capecitabine Citation[54]. Further study should be carried out into newer chemotherapy agents with less toxicity and possibly in combination with targeted therapies.
Targeted therapy
lists examples of targeted agents in MTC. A hallmark of MTC is the association with the proto-oncogene RET, which is involved in cell signaling and regulation of the production of proteins that are essential for spermatogenesis and the development of the autonomic nervous system and kidneys. RET is a transmembrane protein that can be activated by extracellular signals and initiate a complex cascade of intracellular reactions regulating cell growth. A tyrosine kinase enzyme located on the intracellular domain of RET activates this cell signaling cascade. Mutations in specific regions of the RET gene have been described in MTC and these occur in both the sporadic and familial forms of the disease Citation[55,56]. These mutations have motivated experiments looking into corrective gene therapy as a treatment strategy Citation[57].
ZD6474 (Zactima™ [Astra Zeneca], vandetanib) is a novel anilinoquinazoline compound with a molecular weight of 475 Da. It was considered worthy of further development after demonstrating potent inhibition of VEGF receptor (VEGFR)-2 tyrosine kinase in recombinant enzyme assays, as well as additional activity against VEGFR-3, EGF receptor and RET tyrosine kinases. Vandetanib showed excellent selectivity for these kinases compared with related receptor tyrosine kinases, such as PDGF receptor (PDGFR)-β and c-Kit Citation[58–60]. Its activity in hereditary MTC was tested in a Phase II study where 30 patients received vandetanib 300 mg daily. Overall, 20% of subjects achieved a partial response and another 53% had stable disease for more than 24 weeks. The majority of patients (80%) had reductions in their calcitonin levels to less than half the baseline values for at least 4 weeks Citation[61]. It remained to be seen whether this represented a response in the tumor itself or simply a biochemical response to the drug. In a similar Phase II study using a lower dose of the drug (100 mg) as monotherapy in patients with locally advanced or metastatic familial forms of MTC, there were two (11%) responders among the 19 enrolled patients. In this study, disease control was seen in 41% of all patients. Toxicities were manageable in both trials, with the most common adverse events being diarrhea, rash and asymptomatic QTc prolongation on electrocardiogram Citation[62].
The encouraging results of these trials spurred accrual on to an international randomized Phase II trial (known as the ZETA trial) comparing ZD6474 with placebo in patients with inherited and sporadic forms of MTC Citation[63,64]. In this large trial, 331 adults with unresectable locally advanced or metastatic MTC were randomized in a 2:1 manner to receive either ZD6474 (vandetanib) at a dose of 300 mg daily or placebo. Between December 2006 and November 2007, 231 subjects were assigned vandetanib and 100 received placebo. The majority of patients had sporadic disease (90%), metastatic stage (95%) and tumors that were positive for a somatic RET mutation (56%). Patients were followed until disease progression, at which time they were unblinded and had the option to receive vandetanib in an open-label trial; if they chose open-label vandetanib, they were then followed for survival. The median duration of treatment was 90.1 weeks in the vandetanib arm and 39.9 weeks in the placebo arm.
Results at 2-year follow-up showed that 37% of the patients had progression and 15% had died. The primary end point of the study, progression-free survival, was met with the researchers reporting a hazard ratio of 0.46 (95% CI: 0.31–0.69). The median progression-free survival was 19.3 months in the placebo group and had not yet been reached in the vandetanib arm at the time of the presentation at the 14th International Thyroid Congress in 2010. Vandetanib was also associated with statistically significant advantages in secondary end points such as objective response rate (45 vs 13%; odds ratio [OR] = 5.4); disease control rate of 24 weeks or more (OR = 2.64); calcitonin biochemical response (OR = 72.9); CEA biochemical response (OR = 52); and time to worsening of pain (hazard ratio: 0.61). Some of the radiological responses were dramatic, as shown in . In the placebo arm, 12 of 13 responses occurred after the patients had received open-label vandetanib. Adverse events were more common with vandetanib compared with placebo, including diarrhea (56 vs 26%), rash (45 vs 11%), nausea (33 vs 16%), hypertension (32 vs 5%) and headache (26 vs 9%).
Based on these results, Astra Zeneca filed for US FDA approval of the drug in the USA and the European Medicines Agency in Europe late in 2010, receiving approval by the FDA in April 2011 Citation[101].
Further investigation for a medicine with higher affinity for RET led to the development of another TKI inhibitor, XL-184 (cabozantinib), which also blocks VEGFR2 and c-MET. In a Phase I trial including an expansion cohort of 23 subjects with MTC, almost all patients showed some degree of radiographic response, with 12 (55%) out of 22 evaluable patients achieving a partial response and 84% of patients achieving stable disease lasting for more than 3 months. The treatment was well tolerated, with the main side effects including diarrhea, nausea, hypertension and liver function test elevations at a dose of 175 mg daily. Long-term follow-up of these patients revealed a 41% partial response rate in a total of 37 patients in whom the median progression-free survival had not yet been reached Citation[65]. This promising new compound is currently being evaluated in a Phase III study in which patients with unresectable locally advanced or metastatic MTC are randomized 2:1 to receive XL-184 or placebo if they had demonstrated radiologically progressive disease prior to being enrolled Citation[66].
Motesanib
Motesanib (AMG 706), another multikinase inhibitor (VEGF/PDGF receptors, Kit and RET), was studied in a Phase II trial of patients with advanced differentiated thyroid cancer and MTC. All patients received motesanib 125 mg daily until disease progression or unacceptable toxicity. The results for the MTC group (n = 91) after a median follow-up of 49 weeks showed a partial response rate of 2%, according to Response Evaluation Criteria In Solid Tumors (RECIST) criteria. However, 81% had stable disease, which lasted for more than 24 weeks in 48% of patients Citation[67].
Axitinib
Axitinib (AG-013736) is another oral small-molecule tyrosine kinase inhibitor that effectively inhibits VEGFR but not RETCitation[68]. Nevertheless, patients with MTC were included in a multicenter, open-label Phase II study looking at the use of axitinib in advanced or metastatic thyroid carcinoma. Axitinib was administered at a dose of 5 mg twice daily. Of the 60 patients who started therapy, the majority had differentiated thyroid cancer and 18% had MTC. The confirmed partial response rate was 30% by intent-to-treat analysis (31% in differentiated thyroid cancer; 18% in MTC and in one patient with anaplastic thyroid cancer). Responses were seen in patients despite previous treatments with a variety of chemotherapeutic regimens. Median progression-free survival was 18 months. Common adverse events included fatigue, stomatitis, proteinuria, diarrhea, hypertension and nausea Citation[69].
Sorafenib
Sorafenib, a multikinase inhibitor targeting RET and VEGFR, was evaluated in a Phase II trial in patients with advanced MTC, the primary end point of which was objective response. Secondary end points included toxicity assessment and response correlation with tumor markers, functional imaging and RET mutations. Using a two-stage design, 16 or 25 patients were to be enrolled on to arms A (hereditary MTC) and B (sporadic MTC); all patients received sorafenib 400 mg orally twice daily. Only five patients were enrolled in arm A and it was therefore terminated due to slow accrual. Of the 16 patients treated in arm B, one achieved a partial response (6.3%; 95% CI: 0.2–30.2%), 14 had stable disease (87.5%; 95% CI: 61.7–99.5%) and one was nonevaluable. In a post hoc analysis of ten arm B patients with progressive disease before study, one patient had a partial response of 21 months, four patients had stable disease for 15 months, four patients had stable disease for 6 months and one patient had clinical progression of disease. Median progression-free survival was 17.9 months. Common adverse events included diarrhea, hand/foot skin reaction, rash and hypertension. Although serious adverse events were rare, one death was observed. Tumor markers decreased in the majority of patients and RET mutations were detected in ten out of the 12 sporadic MTCs analyzed Citation[70].
Sunitinib
Sunitinib is a multitargeted tyrosine kinase inhibitor of VEGFR and RET, making it a good candidate for a treatment of locally advanced and metastatic MTC. In a Phase II study, 35 patients with thyroid cancer , six of whom had MTC, received 37.5 mg sunitinib daily until progression of disease or unresolved toxicities. The primary end point was overall response rate based on RECIST at the time of maximal response. Secondary end points included evaluation of time to tumor progression, overall survival, and the safety and toxicity of this regimen. Three MTC patients had a partial response, two had progression of disease and one had stable disease. The most common adverse effects were fatigue, diarrhea, hand/foot syndrome and neutropenia. Grade 3 toxicities included cytopenias (46%) diarrhea (17%), hand/foot syndrome (17%) and fatigue (11%). The median survival in this study had not been reached at the time of publication of study results Citation[71].
Expert commentary & five-year view
The diagnosis and treatment of MTC has changed dramatically over the last 50 years. The discovery of RET has allowed insight into the pathogenesis of this disease and has also allowed a chance for detection of the disease at early stages and even to screen family members of the index case to see if they carry the mutation. Early detection of family members carrying a RET mutation allows for the possibility of prophylactic thyroidectomy. The ATA and other authorities have developed guidelines for clinical management, depending on the mutation and family preference. Consensus-based guidelines for definitive surgery have also been developed for patients who have a biopsy-proven diagnosis and excellent survival rates have been achieved for early-stage disease.
For patients with unresectable disease, the field of molecular biology has yielded tyrosine kinase inhibitors such as vandetanib that have shown promising results in randomized studies. The pivotal study with this agent has been reported in abstract form and showed a significant improvement in progression-free survival that had not been reached at the time of the presentation, compared with 19 months with placebo. Other agents with more potent affinity for the RET receptor, such as XL-184, have been developed and are now in clinical trials. The main question for a 5-year view is when should these agents be used and in which patient population. The 10-year survival of patients with metastatic disease is 40%, which is much better than most other solid tumors. The new targeted agents have shown improvement in progression-free survival, but to date there has been no documentation of improved overall survival. The potential toxicities associated with the drug have to be weighed carefully against a disease process such as MTC, which can be indolent and asymptomatic for long periods of time, even when it is progressive.
As new pharmacologic agents are approved for use over the next 5 years, it will be up to the FDA and other authority agencies to determine guidelines for their use. These will probably involve documenting radiographic progression of disease over a nominal time period, as well as symptoms related to the disease. As more agents are developed, hopefully their side-effect profile will improve and the possibility of treating metastatic MTC as a chronic disease similar to hypertension or diabetes may become a reality. It is possible that for some time to come, a combination of novel small-molecule therapies will be necessary to complement surgery, which continues to be the only opportunity for cure. Another novel indication one day might be use of these novel therapies in a neoadjuvant fashion, in order to potentially convert patients with bulky, locally advanced disease to candidates for surgical resection by shrinking their tumors and permitting surgery with acceptable morbidity.
Table 1. American Thyroid Association guidelines on prophylactic thyroidectomy.
Table 2. Targeted agents in medullary thyroid cancer.
Table 3. Ongoing clinical trials in medullary thyroid cancer.
Key issues
• The evolution of medullary thyroid cancer has been dramatic over the past 50 years.
• The discovery of RET has allowed early diagnosis and detection of the disease and allowed prophylactic thyroidectomy to be performed in selected patients.
• Guidelines for prophylactic surgery are based on RET mutations and family preference.
• Definitive surgery for medullary thyroid cancer has resulted in 10-year survival rates of 40% for distant metastatic disease to over 90% for localized disease.
• Targeted therapies against the RET receptor have improved outcomes for patients with metastatic disease.
References
- Carling T, Udelsman R. Cancer of the endocrine system: section 2 thyroid tumors. In: Cancer Principles and Practice. De Vita V, Hellman S, Rosenberg S (Eds). Lippincott Williams and Wilkins, PA, USA, 1503–1521 (2005).
- Ahmed SR, Ball DW. Incidentally discovered medullary thyroid cancer: diagnostic strategies and treatment. J. Clin. Endocrinol. Metab.96, 2359–2367 (2011).
- Hazard JB, Hawk WH, Crile G. Medullary (solid) carcinoma of the thyroid. A clinicopathologic entity. J. Clin. Endocrinol.19, 152–155 (1959).
- Friedel GH, Carey RJ, Rosen H. Familial thyroid cancer. Cancer15, 241–245 (1961).
- Sipple JF. Association of phaeochromocytoma with carcinoma of the thyroid gland. Am. J. Med.31, 163–166 (1961).
- Steiner AL, Goodman AD, Powers SR. Study of a kindred with phaeochromocytoma, hyperparathyroidism and cushings disease; multiple endocrine neoplasia type 2. Medicine47, 371–379 (1968).
- Sakorafas GH, Friess H, Peros G. The genetic basis of hereditary medullary thyroid cancer; clinical implications for the surgeon with a particular emphasis on the role of prophylactic thyroidectomy. Endocr. Relat. Cancer15(4), 871–884 (2008).
- Brandi ML, Gagel RF, Angeli A et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J. Clin. Endocrinol. Metab.86, 5658–5671 (2001).
- Pacini F. Castagna MG, Cipri C et al. Medullary thyroid cancer. Clin. Oncol.22, 475–485 (2010).
- Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer107, 2134–2142 (2006).
- Deshpande HA, Gettinger SN, Sosa JA. Novel chemotherapy for advanced thyroid tumors; small molecules offer great hope. Curr. Opin. Oncol.20, 19–24 (2008).
- Schmimke RN, Hartmann WH. Familial amyloid producing medullary thyroid carcinoma and pheochromocytoma. A distinct genetic entity. Ann. Intern. Med.95, 1027–1039 (1965).
- Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, RET, by DNA rearrangement. Cell42, 581–588 (1985).
- Kloos RT, Eng C, Evans DB et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid19, 565–612 (2009).
- Jiménez C, Hu MI, Gagel RF. Management of medullary thyroid carcinoma. Endocrinol. Metab. Clin. North Am.37(2), 481–496 (2008).
- Grubbs EG, Waguespack SG, Rich TA et al. Do the recent American Thyroid Association (ATA) guidelines accurately guide the timing of prophylactic thyroidectomy in MEN2A? Surgery148, 1302–1310 (2010).
- Breuer C, Tuggle C, Solomon D et al. Pediatric thyroid disease: when is surgery necessary and who should be operating on our children? J. Pediatr. Sci. (In Press) (2011).
- Sosa JA, Bowman HM, Tielsch JM et al. The importance of surgeon experience for clinical and economic outcomes from thyroidectomy. Ann. Surg.228(3), 320–330 (1998).
- Tuggle CT, Roman SA, Wang TS et al. Pediatric endocrine surgery: who is operating on our children? Surgery144(6), 869–877 (2008).
- Kebebew E, Clark OH. Differentiated thyroid cancer: ‘complete’ rational approach. World J. Surg.24(8), 942–951 (2000).
- Thompson GB, Hay ID. Current strategies for surgical management and adjuvant treatment of childhood papillary thyroid carcinoma. World J. Surg.28(12), 1187–1198 (2004).
- Lee JA, Grumbach MM, Clark OH. The optimal treatment for pediatric Graves’ disease is surgery. J. Clin. Endocrinol. Metab.92(3), 801–803 (2007).
- Cooper DS, Doherty GM, Haugen BR et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid19(11), 1167–1214 (2009).
- Tuttle RM, Ball DW, Byrd D et al. Thyroid carcinoma. J. Natl Compr. Canc. Netw.8(11), 1228–1274 (2010).
- Pacini F, Fontanelli M, Fugazzola L et al. Routine measurement of serum calcitonin in nodular thyroid diseases allows the preoperative diagnosis of unsuspected sporadic medullary thyroid carcinoma. J. Clin. Endocrinol. Metab.78, 826–829 (1994).
- Rieu M, Lame MC, Richard A et al. Prevalence of sporadic medullary thyroid carcinoma: the importance of routine measurement of serum calcitonin in the diagnostic evaluation of thyroid nodules. Clin. Endocrinol.42, 453–460 (1995).
- Niccoli P, Wion-Barbot N, Caron P et al. Interest of routine measurement of serum calcitonin: study in a large series of thyroidectomized patients. J. Clin. Endocrinol. Metab.82, 338–341 (1997).
- Vierhapper J, Raber W, Bieglmayer C et al. Routine measurement of plasma calcitonin in nodular thyroid diseases. J. Clin. Endocrinol. Metab.82, 1589–1593 (1997).
- Karanikas G, Moameni A, Poetzi C et al. Frequency and relevance of elevated calcitonin levels in patients with neoplastic and nonneoplastic thyroid disease and in healthy subjects. J. Clin. Endocrinol. Metab.89, 515–519 (2004).
- Vierhapper H, Niederle B, Beiglmayer C et al. Early diagnosis and curative therapy of medullary thyroid carcinoma by routine measurement of serum calcitonin in patients with thyroid disorders. Thyroid15, 1267–1272 (2005).
- Costante G, Meringolo D, Durante C et al. Predictive value of serum calcitonin levels for preoperative diagnosis of medullary thyroid carcinoma in a cohort of 5817 consecutive patients with thyroid nodules. J. Clin. Endocrinol. Metab.92, 450–455 (2007).
- Elisei R, Bottici V, Luchetti F et al. Impact of routine measurement of serum calcitonin on the diagnosis and outcome of medullary thyroid cancer: experience in 10,864 patients with nodular thyroid disorders. J. Clin. Endocrinol. Metab.89, 163–168 (2004).
- Pacini F, Schlumberger M, Dralle H et al. European Thyroid Cancer Taskforce 2006 European consensus for the management of patients with differentiated thyroid carcinoma of the follicular epithelium. Eur. J. Endocrinol.154, 787–803 (2006); erratum 155, 385 (2006).
- Cheung K, Roman SA, Wang TS et al. Calcitonin measurement in the evaluation of thyroid nodules in the United States: a cost–effectiveness and decision analysis. J. Clin. Endocrinol. Metab.93, 2173–2180 (2008).
- Stout N, Rosenberg M, Trentham-Dietz A et al. Retrospective cost-effectiveness analysis of screening mammography. J. Natl Cancer Inst.98, 774–782 (2006).
- Mushlin AI, Fintor L. Is screening for breast cancer cost-effective? Cancer69, 1957–1962 (1992).
- Inodimi JM. Update on the cost–effectiveness of screening for colorectal neoplasia. Curr. Opin. Gastroenterol.19, 44–50 (2003).
- Kim SH, Kim BS, Jung SL et al. Ultrasonographic findings of medullary thyroid carcinoma: a comparison with papillary thyroid carcinoma. Korean J. Radiol.10, 101–105 (2009).
- Lee S, Shin JH, Han BK et al. Medullary thyroid carcinoma: comparison with papillary thyroid carcinoma and application of current sonographic criteria. Am. J. Roentgenol.194, 1090–1094 (2010).
- Papaparaskeva K, Nagel H, Droese M. Cytologic diagnosis of medullary carcinoma of the thyroid gland. Diagn. Cytopathol.22, 351–358 (2000)
- Chang TC, Wu SL, Hsiao YL. Medullary thyroid carcinoma: pitfalls in diagnosis by fine needle aspiration cytology and relationship of cytomorphology to RET proto-oncogene mutations. Acta Cytol.49, 477–482 (2005).
- Bugalho MJ, Santos JR, Sobrinho L. Preoperative diagnosis of medullary thyroid carcinoma: fine needle aspiration cytology as compared with serum calcitonin measurement. J. Surg. Oncol.91, 56–60 (2005).
- Bugalho MJ, Mendonça E, Sobrinho LG. Medullary thyroid carcinoma: an accurate pre-operative diagnosis by reverse transcription-PCR. Eur. J. Endocrinol.143, 335–338 (2000).
- Takano T, Miyauchi A, Matsuzuka F et al. Preoperative diagnosis of medullary thyroid carcinoma by RT-PCR using RNA extracted from leftover cells within a needle used for fine needle aspiration biopsy. J. Clin. Endocrinol. Metab.84, 951–955 (1999).
- Massaro F, Dolcino M, Degrandi R et al. Calcitonin assay in wash-out fluid after fine-needle aspiration biopsy in patients with a thyroid nodule and border-line value of the hormone. J. Endocrinol. Invest.32(4), 308–312 (2009).
- Kudo T, Miyauchi A, Ito Y, Takamura Y, Amino N, Hirokawa M. Diagnosis of medullary thyroid carcinoma by calcitonin measurement in fine-needle aspiration biopsy specimens. Thyroid17(7), 635–638 (2007).
- Boi F, Maurelli I, Pinna G et al. Calcitonin measurement in wash-out fluid from fine needle aspiration of neck masses in patients with primary and metastatic medullary thyroid carcinoma. J. Clin. Endocrinol. Metab.92(6), 2115–2118 (2007).
- Wohllk N, Cote GJ, Bugalho MM et al. Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. J. Clin. Endocrinol. Metab.81, 3740–3745 (1996).
- Machens A, Dralle H. Biomarker-based risk stratification for previously untreated medullary thyroid cancer. J. Clin. Endocrinol. Metab.95, 2655–2663 (2010).
- Modigliani E, Cohen R, Campos JM et al. Prognostic factors for survival and for biochemical cure in medullary thyroid carcinoma: results in 899 patients. Clin. Endocrinol.48, 265–273 (1998).
- Valle LA, Kloos RT. The prevalence of occult medullary thyroid carcinoma at autopsy. J. Clin. Endocrinol. Metab.96(1), E109–E113 (2011).
- Ahuja S, Ernst H. Chemotherapy of thyroid carcinoma. J. Endocrinol. Invest.10(3), 303–310 (1987).
- Matuszcyk A, Petersenn S, Voigt W et al. Chemotherapy with paclitaxel and gemcitabine in progressive medullary and thyroid carcinoma of follicular epithelium. Horm. Metab. Res.42(1), 61–64 (2010).
- Gilliam L, Kohn A, Tasneem L et al. Capecitabine therapy for refractory metastatic thyroid carcinoma; a case series. Thyroid16(8), 801–810 (2006).
- Eng C, Clayton D, Schuffenecker I. The relationship between specific RET protooncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: International RET Mutation Consortium analysis. J. Am. Med. Assoc.27, 1575–1579 (1996).
- Eng C. RET proto-oncogene in the development of human cancer. J. Clin. Oncol.17, 380–393 (1999).
- Messina M, Robinson B. Technology insight: gene therapy and its potential role in the treatment of medullary thyroid carcinoma. Natl Clin. Pract. Endocrinol. Metab.3, 290–301 (2007).
- Wedge SR, Ogilvie DJ, Dukes M et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis and tumor growth following oral administration. Cancer Res.62, 4645–4655 (2002).
- Carlomagno F, Vitagliano D, Guida T et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases: Cancer Res.62, 7284–7290 (2002).
- Herbst RS, Heymach JV, O’Reilly MS et al. Vandetanib (ZD6474): an orally available receptor tyrosine kinase inhibitor that selectively targets pathways critical for tumor growth and angiogenesis. Expert Opin. Investig. Drugs16, 239–249 (2007).
- Wells SA, Gosnell JE, Gagel RF et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J. Clin. Oncol.28, 767–772 (2010).
- Haddad RI, Krebs AD, Vasselli J et al. A phase II open-label study of vandetanib in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J. Clin. Oncol.26, 6024 (2008).
- Wells SA. OC-021. Presented at: 14th International Thyroid Congress. Paris, France, 11–16 September 2010.
- Wells SA, Robinson BG, Gagel RF et al. Vandetanib (VAN) in locally advanced or metastatic medullary thyroid cancer (MTC): a randomized, double-blind Phase III trial (ZETA). J. Clin. Oncol.28(15S), abstract 5503 (2010).
- Kurzrock R, Cohen EE, Sherman SI et al. Long-term results in a cohort of medullary thyroid cancer (MTC) patients (pts) in a Phase I study of XL184 (BMS 907351), an oral inhibitor of MET, VEGFR2, and RET. J. Clin. Oncol.28(15S), 5502 (2010).
- Kurzrock R, Sherman S, Hong D et al. A Phase I study of XL184, a MET, VEGFR2, and RET kinase inhibitor, administered orally to patients (pts) with advanced malignancies including a subgroup of pts with medullary thyroid cancer (MTC). Presented at: 20th EORTCNCI–AACR Symposium on Molecular Targets and Cancer Therapeutics. Geneva, Switzerland, 21–24 October 2008 (Abstract 379).
- Schlumberger MJ, Elisei R, Bastholt L et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer: J. Clin. Oncol.27, 3794–3801 (2009).
- Deshpande HA, Gettinger SN, Sosa JA. Axitinib: the evidence of its potential in the treatment of advanced thyroid cancer. Core Evidence4, 43–48 (2009).
- Cohen EE, Rosen LS, Vokes EE et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a Phase II study. J. Clin. Oncol.26, 4708–4713 (2008).
- Lam ET, Ringel MD, Kloos RT et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J. Clin. Oncol.28(14), 2323–2330 (2010).
- Carr LL, Mankoff DA, Goulart BH et al. Phase II study of daily sunitib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin. Cancer Res.16(21), 5260–5268 (2010).
Websites
- NDA approval letter for vandetanib www.accessdata.fda.gov/drugsatfda_docs/appletter/2011/022405s000ltr.pdf
- Clinicaltrials.gov: Imatinib in Combination With Dacarbazine and Capecitabine in Medullary Thyroid Carcinoma http://clinicaltrials.gov/ct2/show/NCT00354523
- Clinicaltrials.gov: An Efficacy Study Comparing ZD6474 to Placebo in Medullary Thyroid Cancer http://clinicaltrials.gov/ct2/show/NCT00410761
- Clinicaltrials.gov: Efficacy of XL184 (Cabozantinib) in Advanced Medullary Thyroid Cancer (EXAM) http://clinicaltrials.gov/ct2/show/NCT00704730
- Clinicaltrials.gov: An Initial Study of Lithium in Patients With Medullary Thyroid Cancer http://clinicaltrials.gov/ct2/show/NCT00582712
- Clinicaltrials.gov: Everolimus in Treating Patients With Progressive or Recurrent, Unresectable, or Metastatic Thyroid Cancer http://clinicaltrials.gov/ct2/show/NCT01118065
- Clinicaltrials.gov: A Trial Evaluating Single Agent and Combined Efficacy of Pasireotide and Everolimus in Adult Patients With Radioiodine-Refractory Differentiated and Medullary Thyroid Cancer http://clinicaltrials.gov/ct2/show/NCT01270321
Medullary thyroid cancer in the past, present and future: from bench to bedside
To obtain credit, you should first read the journal article. After reading the article, you should be able to answer the following, related, multiple-choice questions. To complete the questions (with a minimum 70% passing score) and earn continuing medical education (CME) credit, please go to www.medscape.org/journal/expertendo. Credit cannot be obtained for tests completed on paper, although you may use the worksheet below to keep a record of your answers. You must be a registered user on Medscape.org. If you are not registered on Medscape.org, please click on the New Users: Free Registration link on the left hand side of the website to register. Only one answer is correct for each question. Once you successfully answer all post-test questions you will be able to view and/or print your certificate. For questions regarding the content of this activity, contact the accredited provider, [email protected]. For technical assistance, contact [email protected]. American Medical Association’s Physician’s Recognition Award (AMA PRA) credits are accepted in the US as evidence of participation in CME activities. For further information on this award, please refer to http://www.ama-assn.org/ama/pub/category/2922.html. The AMA has determined that physicians not licensed in the US who participate in this CME activity are eligible for AMA PRA Category 1 Credits™. Through agreements that the AMA has made with agencies in some countries, AMA PRA credit may be acceptable as evidence of participation in CME activities. If you are not licensed in the US, please complete the questions online, print the AMA PRA CME credit certificate and present it to your national medical association for review.
Activity Evaluation: Where 1 is strongly disagree and 5 is strongly agree
1. You are seeing the parents of a 4-year-old girl referred to you because her 38-year-old mother was recently diagnosed with medullary thyroid cancer (MTC). The only other history of cancer in the family is a case of breast cancer in the patient’s grandmother, which was diagnosed at age 60 years.
The patient’s mother is confused as to why her daughter might be at risk for MTC at such a young age. What can you tell her regarding the epidemiology and prognosis of MTC?
□ A MTC now accounts for half of all cases of thyroid cancer
□ B The incidence of MTC as a proportion of all thyroid cancers is increasing
□ C The vast majority of cases of MTC are familial
□ D The 10-year survival rate of MTC confined to the thyroid gland is 95%
2. After a more focused workup, the young girl is categorized as American Thyroid Association (ATA) risk level A for MTC. Her serum calcitonin and thyroid ultrasound are normal.
What is the best recommendation you can provide to the family regarding prophylactic thyroidectomy for their daughter?
□ A It is probably too late to perform prophylactic thyroidectomy
□ B She should undergo thyroidectomy as soon as possible
□ C A repeat calcitonin level should be performed in 6 months; thyroidectomy should be performed if there is any increase in the calcitonin level
□ D Prophylactic thyroidectomy may be delayed if the parents prefer to avoid surgery
3. Fifteen years later, the patient returns to your clinic with some persistent diarrhea and flushing. Which of the following statements regarding the diagnosis of MTC is most accurate?
□ A Serum calcitonin levels are very specific for MTC
□ B A serum calcitonin level above 50 pg/ml is pathognomonic for MTC
□ C Nodules in MTC are usually smaller and less often cystic compared with papillary thyroid cancer (PTC)
□ D There is significant overlap between ultrasonographic characteristics of MTC and PTC
4. The patient is diagnosed with MTC. Which of the following statements regarding management of MTC is most accurate?
□ A Localized MTC larger than 1.5 cm should not require central neck dissection
□ B Any patient who does not reach a nadir in calcitonin levels at 4 weeks after surgery requires immediate new imaging studies
□ C Vandetanib can improve progression-free survival among patients with locally advanced or metastatic MTC
□ D Response rates for motesanib appear superior to those for vandetanib