Abstract
Pseudoxanthoma elasticum (PXE) is a serious genetic disorder with ectopic mineralization affecting the skin, the eye and the cardiovascular system. The disease is predominantly caused by mutations in the transmembrane ABC protein ABCC6, a putative small substrate transporter. Interestingly, ABCC6 seems virtually absent in the affected organs, whereas a high expression is seen in hepatocytes. This and further published experimental evidence indicate that PXE is a systemic, metabolic liver disease where circulatory changes affect the peripheral mineralization process. Owing to the well-characterized transport of organic substrates by related ABC proteins, it has been proposed that PXE is caused by impaired export of an antimineralization compound to the blood. The authors here present an alternative hypothesis that explains ectopic mineralization in PXE as a consequence of hepatic accumulation of ABCC6 substrate(s) that via gene-regulating effects leads to altered hepatic secretion and activation of antimineralization/anticalcification proteins such as fetuin-A and Gla proteins.
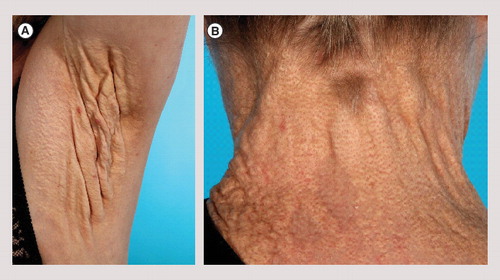
(A) Axillary plaque with severe loss of elasticity in a 56-year-old woman. (B) Late skin symptoms showing coalesced yellow papules forming a plaque surrounding the neck in a 56-year-old woman.
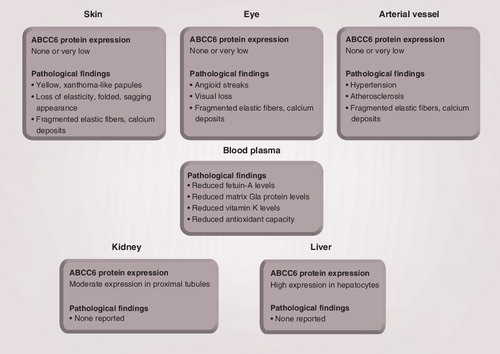
Pseudoxanthoma elasticum-affected tissues like the skin, the eye and the arterial vessel appear devoid of ABCC6 protein. Moderate ABCC6 expression is detected in the kidney, whereas ABCC6 is highly expressed in the liver. No pathological findings have been reported in the kidney and liver of pseudoxanthoma elasticum patients.
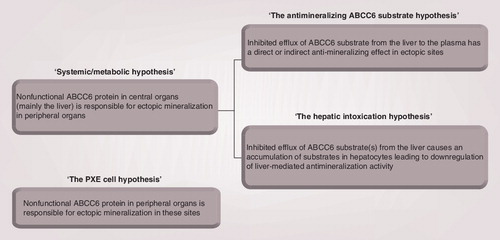
The systemic/metabolic hypothesis of PXE pathogenesis can be divided into two subcategories. ‘The antimineralizing ABCC6 substrate hypothesis’ states that the ABCC6 substrate has antimineralizing properties, and that PXE-related ectopic mineralization is caused by a diminished secretion of this substance from the liver. According to ‘the hepatic intoxication hypothesis’, PXE ectopic mineralization is due to hepatic accumulation of ABCC6 substrate(s) that leads to gene-regulatory changes of hepatic proteins involved in calcification. ‘The PXE cell hypothesis’ states that nonfunctional ABCC6 protein in PXE-affected tissues is the cause of ectopic mineralization in these sites.
PXE: Pseudoxanthoma elasticum.
Pseudoxanthoma elasticum
Pseudoxanthoma elasticum (PXE) is a rare, autosomal recessive disorder characterized by generalized fragmentation and progressive calcification of elastic tissues primarily affecting the skin, the eye and the cardiovascular system. Pathohistological PXE hallmarks include calcium deposits, fragmented elastic fibers and altered proteoglycan composition of elastin-rich tissues. The prevalence of PXE is not fully known because many cases go undiagnosed, but estimates of 1:50,000–1:70,000 have been proposed Citation[1].
The PXE phenotype is rarely detectable until puberty or early adolescence, after which the disease takes a progressive course. Skin papules with a xanthoma-like appearance are early clinical signs of PXE. As the disease progresses, the skin can lose its elasticity and acquire a folded, sagging appearance . Histologically, PXE-affected skin exhibits fragmented and clumped elastic fibers with deposits of calcium Citation[2]. In the eye, PXE manifests as peau d’orange formations and angioid streaks. The latter represents breaks in the elastin-rich Bruch’s membrane of the retina that may eventually lead to visual impairment and central blindness due to hemorrhages and neovascular ingrowth. In the cardiovascular system, calcification of the elastic fibrils in media and intima of arterial vessels may lead to hypertension, angina, general atherosclerosis, myocardial infarction, cerebrovascular insults and gastrointestinal bleeding Citation[3].
The ABCC6 gene: the major disease gene of a multifactorial disease
Several genetic and environmental factors may contribute to PXE development, but the majority of patients exhibit mutations in the ABCC6 gene encoding a putative transporter protein. Mutations in the HBB Citation[4], GGCX Citation[5] and ENPP1 Citation[6] genes may also inflict a PXE-like phenotype. Mutations in these genes are associated with hemoglobinopathies, coagulation defects and arterial calcification, respectively.
ABCC6 is a transmembrane protein that is expressed predominantly in the liver and to a lesser extent in the kidney in humans Citation[7]. Interestingly, PXE-affected tissues seem devoid of ABCC6 protein Citation[7], although low to very low ABCC6 mRNA expression levels were reported in various human tissues Citation[8,9]; however, these observations are conflicting. The prevailing view regarding subcellular protein localization is that ABCC6 is associated with basolateral membranes of human hepatocytes and renal proximal tubule cells Citation[7,10]. A recent study demonstrated that wild-type human ABCC6 protein expressed in mouse hepatocytes located to the plasma membrane, whereas mutated human ABCC6 protein located to the intracellular compartment suggested to be the endoplasmic reticulum Citation[11]. Immunohistochemical studies of human hepatoid carcinomas and normal control liver have reported a canalicular localization of ABCC6 Citation[12]. In the rat liver, both a lateral and a canalicular expression was observed Citation[13–15]. Martin et al. recently reported that ABCC6 locates instead to mitochondrial fractions of mouse hepatocytes fractionated by biochemical methods Citation[16]. Specifically, the protein was shown to localize to sites of dynamic contacts between the outer mitochondrial membrane and the endoplasmic reticulum termed the ‘mitochondria-associated membrane’.
ABCC6 is a member of the ABC protein family that harbors conserved sequences required for ATP hydrolysis. This provides a basis for transport of substances against their concentration gradient. Based on amino acid sequence homology, the ABCC6 protein is classified as a member of the multidrug resistance protein (MRP) subgroup of ABC transporters Citation[17]. ABCC6 is accordingly also termed ‘MRP6’. Within the ABC transporter family, ABCC6 exhibits the highest degree of sequence homology to the ABCC1 protein Citation[17], another MRP transporter that acts as a cellular eliminator of various xenobiotics, organic anions, cobalamin, and so on Citation[18,19]. Based on this observation, it is suggested that ABCC6 also functions as a molecular gate for cellular export. Cellular in vitro transporter assays have, indeed, demonstrated that ABCC6 is capable of transporting anionic glutathione-conjugated compounds Citation[20,21]. Yet endogenously transported ABCC6 substrates and the physiological role of the transporter are obscure. Consequently, the pathomechanism leading to the PXE disease phenotype is largely unknown.
Since the initial reports of mutations in the ABCC6 gene in 2000 Citation[22–25], more than 300 mutations have been identified in PXE patient case studies Citation[26,27]. The majority of these mutations are missense mutations and they are predominantly located in the nucleotide-binding domains of the ABCC6 protein Citation[26,27], which supports the identity of the protein as an ATP-dependent transporter. ABCC6 protein harboring PXE-relevant mutations did, indeed, exhibit reduced transport potential of anionic glutathione-conjugated compounds in an in vitro transporter assay Citation[20].
Studies of knockout mice with an Abcc6−/− genotype have identified aberrant, spontaneous mineralization in tissues similar to those seen in PXE patients including the skin, the retina of the eye and the arterial vessels Citation[28,29]. Like in PXE patients, this mineralization phenotype adopted a progressive course after birth. In addition, connective tissue surrounding the vibrissae displayed profound mineral deposits Citation[29]. Within the connective tissue of the mouse, mineralization was confined to elastic fibers Citation[28,29] and collagen Citation[29]. Mineral imbalance did not, however, appear to be a determinant of these histological findings as serum levels of calcium and phosphorus of knockout mice were similar to those seen in control mice Citation[28,29]. Abcc6-deficient mice generally appear viable and fertile, and their mortality is not significantly different from that of wild-type mice. To date, limited reports on liver and kidney pathophysiology of Abcc6−/− mice have been published. In conclusion, the Abcc6 knockout mouse exhibits mineralization features resembling those of PXE patients, and it underscores the pivotal role of ABCC6 in PXE pathogenesis.
The KK/H1J mouse strain was recently reported to represent a novel PXE animal model Citation[30]. Specifically, this mouse develops mineralization characteristics corresponding to those of the Abcc6 knockout mouse, and genetic analyses have demonstrated an Abcc6 gene mutation causing significantly reduced hepatic Abcc6 protein levels. The mouse strain C3H/HeJ has the same Abcc6 mutation, but the calcification phenotype of this strain is not as severe as that of the KK/H1J strain Citation[30,31].
A zebrafish (Danio rerio) knockdown model represents an alternative system that may be deployed to investigate PXE and ABCC6 transporter function Citation[32]. The study identified two abcc6 genes within the zebrafish genome that were knocked down by morpholino oligonucleotide antisense technology. Knockdown of one of the two genes induced a detectable phenotype of the embryo, which indicates an important role for this abcc6 gene in early zebrafish embryonic development. These embryos did not show evident signs of aberrant mineralization. This might, however, be explained by the fact that zebrafish were studied at their earliest developmental stage, while PXE in humans manifests later in life. A direct clinical relevance of the zebrafish animal model to PXE is not obvious, but it might serve as a tool for further mechanistic studies.
PXE: a systemic/metabolic disorder or a peripheral tissue disease?
The dominating view in the literature is that PXE is a systemic/metabolic disease . This hypothesis was initially raised because ABCC6 protein is expressed primarily in the liver, while PXE clinically manifests in peripheral tissues. This implies that PXE-derived symptoms are probably due to the absence of a circulating factor normally excreted from the liver by ABCC6 Citation[33].
The systemic/metabolic nature of PXE was elegantly consolidated by an animal study in which mineralization symptoms of Abcc6-deficient mice were arrested following parabiotic pairing with healthy wild-type mice Citation[34]. The study demonstrated that blood supply from an unaffected animal was sufficient for phenotypic rescue of a knockout animal. This observation supports the notion that PXE-related mineralization in Abcc6-deficient animals may be elicited by absence of a factor in the blood.
Supporting evidence for ‘the metabolic PXE hypothesis’ is provided by grafting studies with the Abcc6−/− mouse Citation[35]. To determine the contribution of local or metabolic factors to the mineralization phenotype, muzzle skin from a wild-type mouse was grafted onto to the back of an Abcc6-deficient mouse. Although originating from a healthy animal, this graft developed evident signs of mineralization. This observation suggests that the homeostatic milieu is pivotal for the development of aberrant connective tissue mineralization in the PXE mouse model. Likewise, muzzle skin from a knockout mouse that was devoid of mineralization at the time of grafting was grafted onto a wild-type mouse. The fact that this graft sustained a nonmineralized phenotype further underscores that circulating rather than local substances at the site of mineralization influences the mineralization process of PXE Citation[35].
Cellular in vitro experiments confirm the systemic/metabolic hypothesis. In a study of fibroblasts isolated from PXE patients, it was possible to revert the phenotype of these and normal fibroblasts by culturing them in the presence of serum from healthy individuals or from PXE patients Citation[36]. Specifically, the elastic fiber integrity of fibroblasts from healthy individuals was impaired when cultured in PXE serum-containing media. Similarly, PXE fibroblasts exhibited a normal fiber distribution when supplemented with serum from unaffected individuals. Collectively, these observations support the idea that altered levels of one or several factors present in serum account for PXE phenotype development. In line with this notion, the capability of Abcc6−/− knockout mouse serum to influence mineral deposition was investigated in an in vitro calcification assay Citation[37]. This assay is based on aortic smooth muscle cells and allows for mineral precipitation upon addition of inorganic phosphate. Addition of serum from knockout animals to this system resulted in increased calcium deposition when compared with serum from normal mice. Thus, this study suggests that the composition of serum from Abcc6-deficient mice does not sustain complete antimineralizing capacity.
Although the systemic/metabolic hypothesis is widely accepted, the alternative ‘PXE cell hypothesis’ also harbors some support . It postulates that the underlying basis of the disease phenotype is due to impaired ABCC6 function of the affected tissue itself, which results in changes in the local milieu of the affected cells. This view, therefore, assumes that the ABCC6 gene product is expressed at the site of mineralization, although the presence of the protein in peripheral tissues is ambiguous Citation[7,38]. ‘The PXE cell hypothesis’ is based on studies of dermal fibroblasts from PXE patients that exhibit modified cell–cell and cell–matrix properties including increased metalloprotease activity when cultured in vitro Citation[39,40]. Even after several passages in vitro, these cells were reported to sustain the phenotype. This points to intrinsic cellular modifications at the site of clinical manifestations as determinants of the mineralization phenotype.
Investigations of the PXE pathomechanism
Changes in antimineralization activity in plasma & peripheral tissues
Ectopic mineralization in the peripheral tissues of PXE patients is assumed to be attributed to a reduced antimineralization capacity. In fact, fetuin-A, an inhibitor of ectopic mineralization Citation[41], is present at lower levels in serum of the Abcc6−/− mouse and PXE patients Citation[37,42], and it thus seems plausible that a low fetuin-A serum status promotes tissue calcification in PXE. This notion is supported by a study of the Abcc6 knockout mouse in which liver-specific overexpression of a fetuin-A construct increased fetuin-A serum levels and decreased vibrissae-associated calcium deposits Citation[43].
Another factor presumably influencing PXE ectopic mineralization is matrix Gla protein (MGP). This Gla protein requires carboxylation of glutamate residues to exert its function as an inhibitor of local artery calcification Citation[44]. Evidence for a role of MGP as an inducer of PXE calcification includes detection of noncarboxylated, and hence inactive, MGP in the dermis of PXE patients Citation[45] and in the Abcc6 knockout mouse Citation[46]. Moreover, the MGP serum concentration was significantly lower in patient serum Citation[45,47]. It therefore seems probable that an attenuated antimineralization potential by MGP deteriorates PXE mineralization.
Investigations of vitamin K & adenosine as candidate ABCC6 substrates
In 2008, Borst et al. hypothesized that vitamin K or a precursor form of vitamin K is the substrate transported by ABCC6 from the liver into the blood Citation[48]. Vitamin K is essential for γ-carboxylation of glutamate residues in the Gla proteins which, in addition to MGP, include osteocalcin of the bone, coagulation factors involved in blood clotting such as protein C, and so on Citation[49]. According to this hypothesis, a compromised ABCC6 transporter function in PXE patients will result in insufficient vitamin K supply to cells in peripheral tissues. Consequently, the antimineralizing effect of MGP and other mineralization inhibitors is impaired and calcification will ensue Citation[48].
This hypothesis that links local, peripheral vitamin K deficiency and PXE calcification was primed by observations that patients with mutations in the GGCX gene, encoding γ-glutamyl carboxylase (GGCX), presented PXE-like elastic fiber calcification Citation[5]. Hence, it was assumed that an insufficient carboxylation-mediated activation of antimineralization factors contributes to the observed connective tissue calcification in these patients.
The hypothesis was supported by the above-mentioned study of dermis from PXE patients in which it was noticed that the MGP of elastic fibers was almost exclusively present in the noncarboxylated form Citation[45]. MGP would accordingly be inactive and not be capable of preventing elastic fiber mineralization in these patients. A similar pattern was observed in mineralized deposits of the vibrissae of the Abcc6−/− mouse Citation[46]. Furthermore, in line with vitamin K being an ABCC6 substrate, a study reported reduced vitamin K serum levels in PXE patients Citation[50].
Several recent studies with the specific aim to test the vitamin K hypothesis all failed to prove a positive role for vitamin K as a direct determinant of aberrant mineralization in the Abcc6−/− mouse. In three separate animal studies, oral administration of high doses of vitamin K yielded elevated vitamin serum levels, but this elevation was insufficient to counteract ectopic mineralization in kidney arteries Citation[51] and connective tissue surrounding the vibrissae of the Abcc6-deficient mouse Citation[51–53]. These observations were affirmed by intravenous vitamin K administration in which vitamin K supply to peripheral tissue via the liver was bypassed, thus assuring that potentially defective hepatic Abcc6 protein did not affect vitamin K levels in peripheral tissues of Abcc6−/− mice Citation[52]. Altogether, the available data suggest that oral vitamin K supplements cannot ameliorate the calcification phenotype of Abcc6−/− mice.
Furthermore, a physiological role for Abcc6 protein in basolateral hepatic export of vitamin K3 could not be established in a mouse in vivo vitamin K3 liver perfusion experiment Citation[54]. Following perfusion, equal amounts of vitamin K3 were detected in efflux fluid from the liver of Abcc6−/− mice compared with control mice. This suggests that vitamin K is not an Abcc6 substrate, and this notion was supported by in vitro transporter assays Citation[54]. Collectively, vitamin K does, therefore, not appear to be the long-sought ABCC6 substrate.
Adenosine was recently mentioned as another putative ABCC6 substrate. This suggestion was triggered by case studies of patients with loss-of-function mutations in the NT5E gene that exhibited arterial calcification hallmarks similar to those encountered in PXE patients Citation[55,56]. Specifically, elastic fibers of the vessel wall of affected individuals were fragmented. The NT5E gene encodes a 5′-nucleotidase, CD73, that converts AMP into adenosine and inorganic phosphate on the surface of vascular cells. In the case of nonfunctional CD73, a low local vascular adenosine status is associated with increased vessel calcification. The apparent resemblance to the PXE phenotype led to the proposal that adenosine imbalance could play a role in PXE-related ectopic mineralization Citation[55]. It was also hypothesized that adenosine may be a substrate of ABCC6, and that local adenosine paucity governs aberrant mineralization in PXE patients Citation[55].
Data from a vesicular in vitro transporter assay did, however, not point to a role of ABCC6 in adenosine transport Citation[57]. Moreover, in vitro tests of the capacity of adenosine to inhibit mineralization yielded negative results Citation[57]. Based on these results, adenosine does not seem to be a substrate of ABCC6 or to modify the PXE mineralization phenotype.
Is PXE pathology related to oxidative stress?
It was speculated that PXE manifestations could occur due to cellular oxidative stress. This hypothesis was based on cases of β-thalassemia and sickle-cell anemia patients that exhibited a progressive PXE-like mineralization phenotype Citation[4]. Progression of these disorders is, indeed, influenced by oxidative stress Citation[58]. Specifically, angioid streaks and arterial and cutaneous calcification were noted in patients suffering from hemoglobinopathies Citation[4]. Histological changes in dermal specimens from β-thalassemia patients with PXE-like symptoms were similar to the skin alterations of PXE patients Citation[59]. However, these patients were devoid of known PXE-causing mutations Citation[60], and the genetic basis governing clinical findings in β-thalassemia and inherited PXE, therefore, appears distinct. It was recently demonstrated that a β-thalassemia mouse model displayed downregulated Abcc6 protein liver expression Citation[61]. This finding raised the possibility that a reduced level of ABCC6 protein contributes to the mineralization phenotype of humans with β-thalassemia, although this needs further investigation.
A potential link between oxidative imbalance and PXE symptoms finds support in analyses of cultured PXE fibroblasts in which patient cells were reported to display hallmarks of oxidative stress Citation[62,63]. Moreover, oxidative imbalance has been documented in serum from PXE patients Citation[64] and in liver and serum samples of the Abcc6 knockout mouse Citation[65]. If oxidative stress potentiates PXE-derived mineralization, dietary antioxidant supplements may be of particular therapeutic interest as a means of counteracting mineral deposition. To test this possibility, the Abcc6-deficient mouse was fed a diet rich in antioxidants Citation[65]. The capacity for protection against damaging radical species was consequently restored, as evaluated by oxidative stress markers. Despite this cytoprotective recovery, mineralization of the vibrissae, the kidney and the eye was not reduced, which suggests that oxidative stress does not induce connective tissue mineralization Citation[65]. PXE-related oxidative stress may, therefore, be a secondary effect of the disease itself instead of a primary disease modifier. Alternatively, oxidative stress may be a determinant of the severity of clinical PXE outcome, as suggested by a positive correlation between single-nucleotide polymorphisms in genes encoding antioxidant enzymes and early disease onset in PXE patients Citation[66]. In terms of treatment perspective, data from the above-mentioned animal study imply that the proposed beneficial effect of antioxidants in PXE treatment is futile, although this issue needs investigation in a human setting.
‘The hepatic intoxication hypothesis’
Based on the apparent transporter function of ABCC6, its tissue distribution and the pathological findings, the authors now propose an alternative pathomechanism that to a large extent fits with the available experimental data. This ‘hepatic intoxication hypothesis’ is based on the fact that PXE mineralization symptoms are linked to antimineralizating perturbations . According to this hypothesis, the changes in the activity of the antimineralization factors are not due to an impaired ABCC6-mediated secretion of an as yet unknown antimineralization substrate affecting these factors in the plasma. Rather, hepatic accumulation of the ABCC6 substrate (or several substrates), that is not effluxed from the hepatocyte, is responsible for the reduced antimineralization potential. This substrate accumulation may elicit changes in the regulation of genes encoding proteins that directly or indirectly have antimineralizing effects in the blood. Such a permanent imbalance in the mineralization activity may then lead to the peripheral PXE pathology due to ectopic mineralization causing fragmentation of elastic fibers and phenotypic changes of peripheral cells such as fibroblasts. Our hypothesis acknowledges the systemic/metabolic view of the disease in the sense that the underlying defect in ABCC6 is primarily restricted to a central organ, while clinical manifestations occur in peripheral tissues. In fact, symptoms from nonhepatic tissues, in particular the skin, is a common feature of many liver diseases.
The liver-induced pathomechanism of PXE, presented here, finds further support in a recent study by Martin et al. in which genes encoding protein C, ENPP1 and GGCX were reported to be differentially expressed between C3H/HeJ mice that have reduced levels of Abcc6 protein and Abcc6-expressing control mice as evaluated by microarray analyses Citation[16]. Protein C has anticoagulant activity, but also displays gene-regulatory functions Citation[67]. This protein downregulates proinflammatory pathways and it upregulates anti-inflammatory pathways. Like MGP, protein C is a Gla protein and it is probable that a change in the activity of these proteins in PXE is related to the vitamin K-regulated carboxylation of Gla proteins. The enzyme ENPP1 generates pyrophosphate that acts as an inhibitor of calcification and, interestingly, loss-of-function mutations in the gene encoding this protein have been described as the cause of arterial calcification Citation[68]. As described previously, the enzyme GGCX mediates vitamin K-dependent γ-carboxylation of Gla proteins. The available data output from the array study is, however, sparse, and information on whether up- or down-regulation takes place is missing. Nevertheless, the study demonstrates that transcripts – one being involved in calcification – are affected depending on Abcc6 protein status.
The question, therefore, is what kind of ABCC6 substrate could have such an effect on the hepatic antimineralization activity mediated by these genes. At present, it is unknown, but because fetuin-A is a negative acute-phase protein that is downregulated in response to inflammation Citation[69], the authors speculated that a potential ABCC6 substrate possibly has proinflammatory properties. They considered that heme (a component of hemoglobin and heme proteins) could be a substrate of ABCC6 that, if not properly excreted from hepatocytes, could have a slight inflammatory effect because of the well-known proinflammatory effects of heme that stimulate the production of reactive oxygen radicals Citation[70]. Heme was previously mentioned in the context of ABCC6 in relation to investigations of transcriptional regulators of the Abcc6 gene of the mouse Citation[71]. In this study, a transcription factor normally controlling the synthesis of globin and a heme biosynthetic enzyme was identified as a modulator of Abcc6 expression Citation[71]. This link between heme and globin synthesis and Abcc6 expression could point to a role of ABCC6 in heme metabolism.
Heme-induced changes in the antimineralization activity of the liver could also explain the PXE-like symptoms described in patients with hemoglobinopathies such as sickle cell disease and thalassemia. These patients have hemolytic crisis with excessive heme turnover that, in addition to the direct oxidative effects in peripheral tissues, may have intracellular effects in the hepatocytes leading to an imbalance in the mineralization activity controlled by the liver.
The authors have, in fact, tested efflux of radiolabeled hemin [Rasmussen MR et al., Unpublished Data] in HEK-293 cells overexpressing ABCC6 protein. Data from these experiments did not, however, provide experimental evidence for a role of ABCC6 in cellular heme elimination. Although heme may well be involved in PXE pathology in patients with excessive hemolysis, other ABCC6 substrates having regulatory effects on the liver should definitely be considered as causes of classical PXE caused by mutations in ABCC6. The mitochondria-associated membrane, that Abcc6 was recently suggested to be located in Citation[16], is involved in calcium signaling and phospholipid metabolism Citation[72]. Substrates related to these functions should, therefore, be considered as substrates of ABCC6.
Expert commentary
Intensive work, including advanced genetic studies and establishment of animal models, has led to an important insight into the molecular mechanism of PXE. So far, no curative therapy has been developed, but symptomatic treatment may reduce some of the serious symptoms of the disease. Clinical management of PXE patients involves reduction of cardiovascular risk factors such as smoking and hyperlipidemia, and avoidance of drugs that can potentiate bleeding. Due to the risk of ophthalmological complications, awareness of early signs of retinopathy is important. A novel therapy with an antivascular growth factor antibody (ranibizumab or bevacizumab) may delay the serious ophthalmological symptoms Citation[73].
It is of high importance to diagnose PXE patients and to identify the genetic defects causing the disease. Because of the diverse clinical symptoms of PXE, its late onset and overlap with other diseases, many individuals may suffer from undiagnosed PXE. Through genetic screening and clinical inspection by dermatologists and ophthalmologists of relatives of PXE patients with known mutations, it should be possible to identify some of the undiagnosed cases.
Because PXE with high probability is a liver-induced disease, ABCC6 mutational screening of potential liver donors may be relevant. In fact, three cases of acquired PXE in individuals following liver transplantation have been reported Citation[74]. Neither of these patients had mutations in the ABCC6 gene, nor did they present with a family history of PXE. Unfortunately, genetic information of the donors was not available, but it seems reasonable to include ABCC6 genetic screens in liver transplantation procedures.
Five-year view
It seems plausible that the near future will bring major scientific breakthroughs in our understanding of the pathomechanism of PXE. The most outstanding challenge in the field of PXE is to identify the ABCC6 substrate(s). Breakthroughs in this field will most probably give a more complete understanding of the pathological mechanism that may lead to improved treatment and diagnostics of the disease. The field of metabolomics for identification of small molecules in biological samples is rapidly advancing these years in terms of sensitivity, specificity and throughput. This technology appears optimal for identification of substrates of ABC transporters, and metabolomic programs have already been initiated to search for ABC substrates Citation[75–77]. By comparing liver extracts, blood and other biological samples from PXE patients and animals with experimental PXE to appropriate controls, it seems reasonable to suggest that important progress can be made in terms of substrate identification within the near future. Furthermore, a clue to the identity of ABCC6 substrates may be provided by thorough microarray analyses of biological samples from PXE patients and the available experimental animal models.
The Abcc6−/− mouse may be an ideal platform for analysis of the involvement of liver (and kidney) in PXE pathogenesis to further test ‘the hepatic intoxication hypothesis’ presented here. Specifically, grafting of liver and kidneys from healthy mice to Abcc6-deficient mice and vice versa may elucidate the importance of each organ in the mineralization phenotype development. Likewise, selective expression of Abcc6 in either organ of the Abcc6−/− knockout mouse may address the issue of contribution of hepatic and renal ABCC6 protein expression to PXE pathology Citation[10].
If the ABCC6 substrate is identified, medicine, already on the market, may be suitable to change the effects of the decreased transport of the substrate(s). If novel medicine has to be developed, the time for implementation of a new effective therapy may take much longer. Without knowing the substrate identity, new medicine may also be developed within a few years by identifying agents that inflict with the mineralization process in the blood. In this line, several studies have recently presented intriguing data showing that a magnesium-rich diet reduces the peripheral mineralization deposits and PXE pathology in the PXE mouse model Citation[78–82].
Key issues
• Pseudoxanthoma elasticum (PXE) is a hereditary disease of ectopic mineralization affecting connective tissues of primarily the skin, the eye and the cardiovascular system.
• The majority of PXE patients display mutations in the ABCC6 gene encoding a putative transporter of unknown function that is expressed mainly in the liver. Cases of patients with PXE symptoms and mutations in genes besides ABCC6 have been reported.
• Studies of a PXE mouse model have demonstrated that the nature of the disorder is systemic/metabolic, yet the pathomechanism remains obscure. The predominant view on this issue is that nonfunctional ABCC6 in the liver prevents export of a compound with antimineralization properties to the blood, which leads to ectopic mineralization in peripheral tissues.
• The authors propose an alternative hypothesis explaining the overall pathomechanism of PXE: ‘the hepatic intoxication hypothesis’. According to this hypothesis, hepatic accumulation of the ABCC6 substrate (or several substrates) induces gene-regulatory events including downregulation of genes encoding proteins with direct or indirect antimineralization effects.
• Identification of the as yet unknown ABCC6 substrate(s) will be a major breakthrough in the understanding of PXE pathogenesis that may facilitate the development of new therapy for PXE patients.
Financial & competing interests disclosure
The authors were supported by The Novo Nordisk Foundation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Uitto J, Li Q, Jiang Q. Pseudoxanthoma elasticum: molecular genetics and putative pathomechanisms. J. Invest. Dermatol. 130(3), 661–670 (2010).
- Plomp AS, Toonstra J, Bergen AA, van Dijk MR, de Jong PT. Proposal for updating the pseudoxanthoma elasticum classification system and a review of the clinical findings. Am. J. Med. Genet. A 152A(4), 1049–1058 (2010).
- Kornet L, Bergen AA, Hoeks AP et al. In patients with pseudoxanthoma elasticum a thicker and more elastic carotid artery is associated with elastin fragmentation and proteoglycans accumulation. Ultrasound Med. Biol. 30(8), 1041–1048 (2004).
- Aessopos A, Farmakis D, Loukopoulos D. Elastic tissue abnormalities resembling pseudoxanthoma elasticum in β thalassemia and the sickling syndromes. Blood 99(1), 30–35 (2002).
- Vanakker OM, Martin L, Gheduzzi D et al. Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. J. Invest. Dermatol. 127(3), 581–587 (2007).
- Nitschke Y, Baujat G, Botschen U et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am. J. Hum. Genet. 90(1), 25–39 (2012).
- Scheffer GL, Hu X, Pijnenborg AC, Wijnholds J, Bergen AA, Scheper RJ. MRP6 (ABCC6) detection in normal human tissues and tumors. Lab. Invest. 82(4), 515–518 (2002).
- Kool M, van der Linden M, de Haas M, Baas F, Borst P. Expression of human MRP6, a homologue of the multidrug resistance protein gene MRP1, in tissues and cancer cells. Cancer Res. 59(1), 175–182 (1999).
- Beck K, Hayashi K, Dang K, Hayashi M, Boyd CD. Analysis of ABCC6 (MRP6) in normal human tissues. Histochem. Cell Biol. 123(4–5), 517–528 (2005).
- Uitto J, Bercovitch L, Terry SF, Terry PF. Pseudoxanthoma elasticum: progress in diagnostics and research towards treatment: summary of the 2010 PXE International Research Meeting. Am. J. Med. Genet. A 155A(7), 1517–1526 (2011).
- Le Saux O, Fülöp K, Yamaguchi Y et al. Expression and in vivo rescue of human ABCC6 disease-causing mutants in mouse liver. PLoS ONE 6(9), e24738 (2011).
- Kamata S, Kishimoto T, Kobayashi S, Miyazaki M. Expression and localization of ATP binding cassette (ABC) family of drug transporters in gastric hepatoid adenocarcinomas. Histopathology 52(6), 747–754 (2008).
- Madon J, Hagenbuch B, Landmann L, Meier PJ, Stieger B. Transport function and hepatocellular localization of mrp6 in rat liver. Mol. Pharmacol. 57(3), 634–641 (2000).
- Gao B, St Pierre MV, Stieger B, Meier PJ. Differential expression of bile salt and organic anion transporters in developing rat liver. J. Hepatol. 41(2), 201–208 (2004).
- Donner MG, Warskulat U, Saha N, Häussinger D. Enhanced expression of basolateral multidrug resistance protein isoforms Mrp3 and Mrp5 in rat liver by LPS. Biol. Chem. 385(3–4), 331–339 (2004).
- Martin LJ, Lau E, Singh H et al. ABCC6 localizes to the mitochondria-associated membrane. Circ. Res. 111(5), 516–520 (2012).
- Belinsky MG, Kruh GD. MOAT-E (ARA) is a full-length MRP/cMOAT subfamily transporter expressed in kidney and liver. Br. J. Cancer 80(9), 1342–1349 (1999).
- Slot AJ, Molinski SV, Cole SP. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 50(1), 179–207 (2011).
- Beedholm-Ebsen R, Van de Wetering K, Hardlei T, Nexø E, Borst P, Moestrup SK. Identification of multidrug resistance protein 1 (MRP1/ABCC1) as a molecular gate for cellular export of cobalamin. Blood 115(8), 1632–1639 (2010).
- Iliás A, Urbán Z, Seidl TL et al. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6). J. Biol. Chem. 277(19), 16860–16867 (2002).
- Belinsky MG, Chen ZS, Shchaveleva I, Zeng H, Kruh GD. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6). Cancer Res. 62(21), 6172–6177 (2002).
- Bergen AA, Plomp AS, Schuurman EJ et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 25(2), 228–231 (2000).
- Le Saux O, Urban Z, Tschuch C et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 25(2), 223–227 (2000).
- Struk B, Cai L, Zäch S et al. Mutations of the gene encoding the transmembrane transporter protein ABC-C6 cause pseudoxanthoma elasticum. J. Mol. Med. 78(5), 282–286 (2000).
- Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J. Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc. Natl Acad. Sci. USA 97(11), 6001–6006 (2000).
- Miksch S, Lumsden A, Guenther UP et al. Molecular genetics of pseudoxanthoma elasticum: type and frequency of mutations in ABCC6. Hum. Mutat. 26(3), 235–248 (2005).
- Pfendner EG, Vanakker OM, Terry SF et al. Mutation detection in the ABCC6 gene and genotype–phenotype analysis in a large international case series affected by pseudoxanthoma elasticum. J. Med. Genet. 44(10), 621–628 (2007).
- Gorgels TG, Hu X, Scheffer GL et al. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum. Mol. Genet. 14(13), 1763–1773 (2005).
- Klement JF, Matsuzaki Y, Jiang QJ et al. Targeted ablation of the abcc6 gene results in ectopic mineralization of connective tissues. Mol. Cell. Biol. 25(18), 8299–8310 (2005).
- Li Q, Berndt A, Guo H, Sundberg JP, Uitto J. A novel animal model for pseudoxanthoma elasticum: the KK/HlJ mouse. Am. J. Pathol. 181(4), 1190–1196 (2012).
- Aherrahrou Z, Doehring LC, Ehlers EM et al. An alternative splice variant in Abcc6, the gene causing dystrophic calcification, leads to protein deficiency in C3H/He mice. J. Biol. Chem. 283(12), 7608–7615 (2008).
- Li Q, Sadowski S, Frank M et al. The abcc6a gene expression is required for normal zebrafish development. J. Invest. Dermatol. 130(11), 2561–2568 (2010).
- Uitto J, Pulkkinen L, Ringpfeil F. Molecular genetics of pseudoxanthoma elasticum: a metabolic disorder at the environment–genome interface? Trends Mol. Med. 7(1), 13–17 (2001).
- Jiang Q, Oldenburg R, Otsuru S, Grand-Pierre AE, Horwitz EM, Uitto J. Parabiotic heterogenetic pairing of Abcc6-/-/Rag1-/- mice and their wild-type counterparts halts ectopic mineralization in a murine model of pseudoxanthoma elasticum. Am. J. Pathol. 176(4), 1855–1862 (2010).
- Jiang Q, Endo M, Dibra F, Wang K, Uitto J. Pseudoxanthoma elasticum is a metabolic disease. J. Invest. Dermatol. 129(2), 348–354 (2009).
- Le Saux O, Bunda S, Van Wart CM et al. Serum factors from pseudoxanthoma elasticum patients alter elastic fiber formation in vitro. J. Invest. Dermatol. 126(7), 1497–1505 (2006).
- Jiang Q, Li Q, Uitto J. Aberrant mineralization of connective tissues in a mouse model of pseudoxanthoma elasticum: systemic and local regulatory factors. J. Invest. Dermatol. 127(6), 1392–1402 (2007).
- Hu X, Peek R, Plomp A et al. Analysis of the frequent R1141X mutation in the ABCC6 gene in pseudoxanthoma elasticum. Invest. Ophthalmol. Vis. Sci. 44(5), 1824–1829 (2003).
- Quaglino D, Boraldi F, Barbieri D, Croce A, Tiozzo R, Pasquali Ronchetti I. Abnormal phenotype of in vitro dermal fibroblasts from patients with pseudoxanthoma elasticum (PXE). Biochim. Biophys. Acta 1501(1), 51–62 (2000).
- Quaglino D, Sartor L, Garbisa S et al. Dermal fibroblasts from pseudoxanthoma elasticum patients have raised MMP-2 degradative potential. Biochim. Biophys. Acta 1741(1–2), 42–47 (2005).
- Ketteler M, Bongartz P, Westenfeld R et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet 361(9360), 827–833 (2003).
- Hendig D, Schulz V, Arndt M, Szliska C, Kleesiek K, Götting C. Role of serum fetuin-A, a major inhibitor of systemic calcification, in pseudoxanthoma elasticum. Clin. Chem. 52(2), 227–234 (2006).
- Jiang Q, Dibra F, Lee MD, Oldenburg R, Uitto J. Overexpression of fetuin-A counteracts ectopic mineralization in a mouse model of pseudoxanthoma elasticum (abcc6(-/-)). J. Invest. Dermatol. 130(5), 1288–1296 (2010).
- Luo G, Ducy P, McKee MD et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 386(6620), 78–81 (1997).
- Gheduzzi D, Boraldi F, Annovi G et al. Matrix Gla protein is involved in elastic fiber calcification in the dermis of pseudoxanthoma elasticum patients. Lab. Invest. 87(10), 998–1008 (2007).
- Li Q, Jiang Q, Schurgers LJ, Uitto J. Pseudoxanthoma elasticum: reduced γ-glutamyl carboxylation of matrix GLA protein in a mouse model (Abcc6-/-). Biochem. Biophys. Res. Commun. 364(2), 208–213 (2007).
- Hendig D, Zarbock R, Szliska C, Kleesiek K, Götting C. The local calcification inhibitor matrix Gla protein in pseudoxanthoma elasticum. Clin. Biochem. 41(6), 407–412 (2008).
- Borst P, Van de Wetering K, Schlingemann R. Does the absence of ABCC6 (multidrug resistance protein 6) in patients with pseudoxanthoma elasticum prevent the liver from providing sufficient vitamin K to the periphery? Cell Cycle 7(11), 1575–1579 (2008).
- Booth SL. Roles for vitamin K beyond coagulation. Annu. Rev. Nutr. 29, 89–110 (2009).
- Vanakker OM, Martin L, Schurgers LJ et al. Low serum vitamin K in PXE results in defective carboxylation of mineralization inhibitors similar to the GGCX mutations in the PXE-like syndrome. Lab. Invest. 90(6), 895–905 (2010).
- Gorgels TG, Waarsing JH, Herfs M et al. Vitamin K supplementation increases vitamin K tissue levels but fails to counteract ectopic calcification in a mouse model for pseudoxanthoma elasticum.J. Mol. Med. 89(11), 1125–1135 (2011).
- Jiang Q, Li Q, Grand-Pierre AE, Schurgers LJ, Uitto J. Administration of vitamin K does not counteract the ectopic mineralization of connective tissues in Abcc6 (-/-) mice, a model for pseudoxanthoma elasticum. Cell Cycle 10(4), 701–707 (2011).
- Brampton C, Yamaguchi Y, Vanakker O et al. Vitamin K does not prevent soft tissue mineralization in a mouse model of pseudoxanthoma elasticum. Cell Cycle 10(11), 1810–1820 (2011).
- Fülöp K, Jiang Q, Van de Wetering K et al. ABCC6 does not transport vitamin K3–glutathione conjugate from the liver: relevance to pathomechanisms of pseudoxanthoma elasticum. Biochem. Biophys. Res. Commun. 415(3), 468–471 (2011).
- Markello TC, Pak LK, St Hilaire C et al. Vascular pathology of medial arterial calcifications in NT5E deficiency: implications for the role of adenosine in pseudoxanthoma elasticum. Mol. Genet. Metab. 103(1), 44–50 (2011).
- St Hilaire C, Ziegler SG, Markello TC et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 364(5), 432–442 (2011).
- Szabó Z, Váradi A, Li Q, Uitto J. ABCC6 does not transport adenosine – relevance to pathomechanism of pseudoxanthoma elasticum. Mol. Genet. Metab. 104(3), 421; author reply 422 (2011).
- Chan AC, Chow CK, Chiu D. Interaction of antioxidants and their implication in genetic anemia. Proc. Soc. Exp. Biol. Med. 222(3), 274–282 (1999).
- Baccarani-Contri M, Bacchelli B, Boraldi F et al. Characterization of pseudoxanthoma elasticum-like lesions in the skin of patients with β-thalassemia. J. Am. Acad. Dermatol. 44(1), 33–39 (2001).
- Hamlin N, Beck K, Bacchelli B, Cianciulli P, Pasquali-Ronchetti I, Le Saux O. Acquired pseudoxanthoma elasticum-like syndrome in β-thalassaemia patients. Br. J. Haematol. 122(5), 852–854 (2003).
- Martin L, Douet V, VanWart CM, Heller MB, Le Saux O. A mouse model of β-thalassemia shows a liver-specific down-regulation of Abcc6 expression. Am. J. Pathol. 178(2), 774–783 (2011).
- Pasquali-Ronchetti I, Garcia-Fernandez MI, Boraldi F et al. Oxidative stress in fibroblasts from patients with pseudoxanthoma elasticum: possible role in the pathogenesis of clinical manifestations. J. Pathol. 208(1), 54–61 (2006).
- Boraldi F, Annovi G, Guerra D et al. Fibroblast protein profile analysis highlights the role of oxidative stress and vitamin K recycling in the pathogenesis of pseudoxanthoma elasticum. Proteomics. Clin. Appl. 3(9), 1084–1098 (2009).
- Garcia-Fernandez MI, Gheduzzi D, Boraldi F et al. Parameters of oxidative stress are present in the circulation of PXE patients. Biochim. Biophys. Acta 1782(7–8), 474–481 (2008).
- Li Q, Jiang Q, Uitto J. Pseudoxanthoma elasticum: oxidative stress and antioxidant diet in a mouse model (Abcc6-/-). J. Invest. Dermatol. 128(5), 1160–1164 (2008).
- Zarbock R, Hendig D, Szliska C, Kleesiek K, Götting C. Pseudoxanthoma elasticum: genetic variations in antioxidant genes are risk factors for early disease onset. Clin. Chem. 53(10), 1734–1740 (2007).
- Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood 109(8), 3161–3172 (2007).
- Rutsch F, Ruf N, Vaingankar S et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat. Genet. 34(4), 379–381 (2003).
- Lebreton JP, Joisel F, Raoult JP, Lannuzel B, Rogez JP, Humbert G. Serum concentration of human α 2 HS glycoprotein during the inflammatory process: evidence that α 2 HS glycoprotein is a negative acute-phase reactant. J. Clin. Invest. 64(4), 1118–1129 (1979).
- Sadrzadeh SM, Graf E, Panter SS, Hallaway PE, Eaton JW. Hemoglobin. A biologic Fenton reagent. J. Biol. Chem. 259(23), 14354–14356 (1984).
- Douet V, VanWart CM, Heller MB, Reinhard S, Le Saux O. HNF4α and NF-E2 are key transcriptional regulators of the murine Abcc6 gene expression. Biochim. Biophys. Acta 1759(8–9), 426–436 (2006).
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 19(2), 81–88 (2009).
- Finger RP, Charbel Issa P, Hendig D, Scholl HP, Holz FG. Monthly ranibizumab for choroidal neovascularizations secondary to angioid streaks in pseudoxanthoma elasticum: a one-year prospective study. Am. J. Ophthalmol. 152(4), 695–703 (2011).
- Bercovitch L, Martin L, Chassaing N et al. Acquired pseudoxanthoma elasticum presenting after liver transplantation. J. Am. Acad. Dermatol. 64(5), 873–878 (2011).
- Van de Wetering K, Feddema W, Helms JB, Brouwers JF, Borst P. Targeted metabolomics identifies glucuronides of dietary phytoestrogens as a major class of MRP3 substrates in vivo. Gastroenterology 137(5), 1725–1735 (2009).
- Krumpochova P, Sapthu S, Brouwers JF et al. Transportomics: screening for substrates of ABC transporters in body fluids using vesicular transport assays. FASEB J. 26(2), 738–747 (2012).
- Van de Wetering K, Sapthu S. ABCG2 functions as a general phytoestrogen sulfate transporter in vivo. FASEB J. 26(10), 4014–4024 (2012).
- Li Q, Larusso J, Grand-Pierre AE, Uitto J. Magnesium carbonate-containing phosphate binder prevents connective tissue mineralization in Abcc6(-/-) mice–potential for treatment of pseudoxanthoma elasticum. Clin. Transl. Sci. 2(6), 398–404 (2009).
- LaRusso J, Li Q, Jiang Q, Uitto J. Elevated dietary magnesium prevents connective tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6(-/-)). J. Invest. Dermatol. 129(6), 1388–1394 (2009).
- Gorgels TG, Waarsing JH, de Wolf A, ten Brink JB, Loves WJ, Bergen AA. Dietary magnesium, not calcium, prevents vascular calcification in a mouse model for pseudoxanthoma elasticum. J. Mol. Med. 88(5), 467–475 (2010).
- Jiang Q, Uitto J. Restricting dietary magnesium accelerates ectopic connective tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6(-/-)). Exp. Dermatol. 21(9), 694–699 (2012).
- Kupetsky-Rincon EA, Li Q, Uitto J. Magnesium reduces carotid intima-media thickness in a mouse model of pseudoxanthoma elasticum: a novel treatment biomarker. Clin. Transl. Sci. 5(3), 259–264 (2012).