Hb: Hemoglobin; ICAM: Intercellular adhesion molecule; RBC: Red blood cell; VCAM: Vascular cell adhesion molecule.
Sickle cell disease (SCD) is characterized by chronic hemolysis and intermittent vaso-occlusion, and transfusion is indicated for certain acute complications, such as acute chest syndrome, severe anemia, stroke and splenic sequestration. Hyperhemolysis syndrome (HS) is a serious and potentially life-threatening complication of red blood cell (RBC) transfusion and has been well-described in pediatric and adult SCD patients. A case of fatal HS has been reported in a child with SCD Citation[1].
Presenting features of HS include Citation[2–8]:
• Fever and sickle pain (common presenting features)
• Development of severe anemia after transfusion: post-transfusion hemoglobin (Hb) level lower than the pretransfusion value
• Evidence of hemolysis (hemoglobinuria, hyperbilirubinemia and raised lactate dehydrogenase [LDH])
• A fall in absolute reticulocyte count (decrease from patient’s usual base level)
• Recovery manifested by a rise in Hb and reticulocyte count
• Additional transfusion may exacerbate ongoing hemolysis (may lead to protracted course or even death)
• Further classified into acute and delayed forms
• HS may recur following subsequent transfusion (although this is extremely rare)
Hyperhemolysis syndrome has also been reported in patients with underlying myelofibrosis Citation[9], thalassemia Citation[10] and anemia of chronic disorder Citation[11].
The term HS is used because the post-transfusion Hb level is lower than the pretransfusion value, suggesting destruction of both the patient’s own RBCs and transfused RBCs. This differs from the conventional delayed hemolytic transfusion reactions (DHTRs). An anamnestic immune response to an antigen carried by the transfused cells constitutes a pre-requisite for the occurrence of DHTR and anemia is mainly due to short survival of the transfused red cells.
Hyperhemolysis syndrome can be further classified into acute and delayed forms Citation[6,12]. The acute form usually occurs less than 7 days after receiving the blood transfusion. The direct antiglobulin test (DAT) is usually negative and no red cell alloantibodies are identified in the patient’s serum. In cases with preformed alloantibodies detected in the patient’s serum, transfusion of antigen-negative crossmatch-compatible blood may not prevent the occurrence of HS. In the acute form of HS, serological investigations of post-transfusion and follow-up samples may not reveal the formation of new/additional RBC alloantibodies (i.e., there is no evidence of RBC antibody-mediated hemolysis).
The delayed form usually occurs more than 7 days post-transfusion. The DAT is positive and new alloantibodies are often identified in the patient’s serum sample post-tranfusion.
Pathogenesis
The pathogenesis of HS remains unclear but appears to be complex as the Hb level decreases below the pretransfusion level. In the acute form of HS, RBC alloantibodies are not detected in the patient’s samples and HS may still occur despite providing ABO, Rhesus- and K-matched crossmatch-compatible RBC units Citation[2,3]. Absolute reticulocytosis is generally presented as a mechanism that is compensatory for hemolysis in steady-state SCD. However, in HS, a fall in reticulocyte count during hemolysis was recorded and recovery manifested by a rise in reticulocyte count. There are conflicting opinions reported in the literature as to whether the significant decrease in Hb results from hemolysis of recipient cells or is due to ineffective erythropoiesis; characterized by reticulocytopenia Citation[2,6,8].
The possible mechanisms include bystander hemolysis Citation[8], suppression of erythropoiesis and RBCs being destroyed by activated macrophages Citation[6].
Bystander hemolysis
King et al. suggested the bystander hemolysis mechanism Citation[8]. King’s group have reported on five patients who presented with HS after receiving exchange transfusion, of which three were delayed and two were acute forms. They conducted serial measurements of HbA and HbS levels in the peripheral blood as a marker to document both the presence and absence of autologous cell destruction. In two of the cases, the authors concluded that the cells were destroyed by bystander hemolysis. The term bystander hemolysis was first coined by Petz and Garraty to describe the phenomenon of “immune hemolysis of cells that are negative for the antigen against which the relevant antibody is directed” Citation[13]. Garraty also suggested that in HS, in the absence of RBC alloantibodies, other antibodies reacting with transfused foreign antigens (e.g., HLA and plasma proteins) may cause complement activation, leading to bystander hemolysis Citation[14]. In two of the cases described by King et al., no RBC alloantibodies were identified and HLA antibodies were not investigated. Therefore, it is difficult to prove the actual nature of the reaction that contributed to bystander hemolysis in these cases.
Suppression of erythropoiesis
Association of reticulocytopenia (a significant decrease from the patient’s usual absolute reticulocyte level) was first described by Petz et al.Citation[2]. They reported four delayed and one acute form of HS and suggested that the apparent increased rate of the hemolysis of autologous RBC was partly due to transfusion-induced suppression of erythropoiesis. It is interesting to note that each patient received an additional transfusion of 7–18 units (mean: 13 units) during the course and were discharged on days 22, 24, 29, 36 and 52 after admission. As these patients received multiple transfusions over a long period, it is possible that the suppression of erythropoiesis might play some part in contributing to the worsening anemia in this group. Two patients were given oral steroids and both recovered gradually. The response to steroids suggests that another factor other than suppression of hemopoiesis may be involved in the pathogenesis.
Activated macrophages
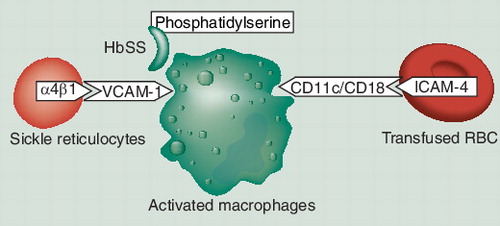
Sickle cell disease is characterized by intravascular and extravascular hemolysis, and destruction of sickle cells may occur at a fairly substantial pace. Abnormal rigidity of the cells and the unusual tendency of HbSS cells to adhere to macrophages play an important role in short RBC survival.
As RBC antibodies are not detected in the acute form, we have previously proposed that the patient’s own cells and transfused cells were destroyed by macrophages. Intravenous immunoglobulin (IVIG) and steroids both have suppressive actions on macrophages; we have also recommended that treatment with IVIG and steroids should be considered in cases presenting with rapid severe life-threatening HS. We have previously reported six cases (five acute and one delayed form) Citation[3–6]. One of the acute cases presented with a recurrent episode of HS Citation[6]. Nadir Hb levels for these cases were 4.8, 4.4, 3, 3.8 and 3.5 g/dl, and for the recurrent case, the nadir Hb levels were 4.1 and 3.2 g/dl for the first and second episodes, respectively. During the first episode, the child responded to IVIG–steroid therapy and did not need blood. However, for the second episode, the child was given additional transfusion with IVIG–intravenous steroid therapy. All of the remaining four patients with the acute form of HS did receive additional transfusion with IVIG–intravenous steroid therapy. In the delayed form, the patient responded to IVIG–intravenous steroid therapy and transfusion was avoided Citation[5].
Evidence of destruction of patients’ own cells & transfused cells
Serial measurement of high-performance liquid chromatography (HPLC) analysis of the urine was conducted in one of our cases. The HPLC system is an analytic procedure that measures the relative proportion of differential Hbs in the urine sample. HPLC documented the presence of HbA and HbS in the urine. The findings of serial measurement of RBC Hb electrophoresis Citation[8] and HPLC analysis of the urine support the fact that the decrease in Hb in HS was linked to the destruction/hemolysis of autologous and transfused cells. This was the first reported case using urine HPLC analysis to confirm the evidence of hemolysis Citation[4].
Causes of the fall & rise of reticulocyte count
Reticulocytopenia was documented at the onset of hemolysis in five of our six reported cases, and HS occurred in the cases after receiving 2, 3, 3, 3, 4 and 2 RBC units (mean: 3 units). It is unlikely that receiving transfusion of 2–3 RBC units may lead to suppression of erythropoiesis. Bone marrow aspiration was conducted in one of the cases due to persistent reticulocytopenia despite receiving erythropoietin for 20 days and oral steroids for 14 days. Marrow aspirate showed erythroid hyperplasia with erythroblasts at all stages of maturation. This was the only reported case whereby bone marrow aspirate was examined during HS in SCD Citation[4].
An increase in reticulocyte count together with Hb level in response to IVIG–steroid treatment was recorded in all of our reported cases Citation[3–6]. Therefore, we have concluded that the reticulocytopenia in HS is not due to suppression of erythropoiesis but is probably due to peripheral consumption (i.e., destroyed by macrophages) Citation[4].
HbSS/reticulocyte adhesion with macrophages
Macrophages are already activated in patients with SCD Citation[15]. Fever and pain are the common presenting symptoms in HS, and these inflammation and immune responses might further activate macrophages.
Hemoglobin S cells adhere to macrophages more readily than HbA cells and this is due to exposure of aminophosphatides (phosphatidylserine) in the outer membrane of the sickle RBC Citation[16].
Sickle reticulocytes adhere via α4β1 VCAM-1 to the vascular endothelium, and reticulocytes are emerging as key participants in the pathophysiology of SCD vascular occlusion Citation[17]. VCAM-1 is not confined to the endothelium and is also expressed on macrophages. Therefore, reticulocytes might also bind to macrophages via the same α4β1 adhesion mechanism. We have previously suggested that this increased adherence of HbSS/reticulocytes to macrophages may play an important role in the destruction of autologous cells, resulting in reticulocytopenia and anemia Citation[3–6].
HbAA adhesion with macrophages
Recent studies have demonstrated that ICAM-4, a glycoprotein expressed on RBCs and erythroid precursor cells, interacts with macrophages via integrin receptors CD11c–CD18. Inhibition of erythrophagocytosis by anti-ICAM-4 and anti-integrin antibodies supports the role of contact lysis of RBCs by macrophages Citation[18].
In addition, further evidence is emerging that the autologous cells (HbSS/reticulocytes) and transfused cells (HbAA) can also interact with macrophages via different adhesion mechanism.
Acute form of HS
In the acute form of HS, both autologous cells and transfused cells are destroyed in the absence of red cell alloantibodies. Therefore, we have concluded that the patient’s RBCs and transfused RBCs are destroyed by activated macrophages, both by contact lysis and erythrophagocytosis Citation[6].
Delayed form of HS: bystander mechanism
In the delayed form of HS, new RBC alloantibodies are often identified in post-transfusion patients’ samples. Darbi et al. have reported a delayed form of HS in a case of anemia of chronic disorder due to anti-K. The patient experienced ongoing hemolysis for 9 weeks, despite receiving 19 units of K-cross-matched compatible units. This case represents a typical example of the bystander hemolytic mechanism Citation[11]. It also demonstrates that continued transfusion prolongs the course of the hemolytic process.
Possible role of HLA antibody/activated macrophages in HS
HLA antibody formation is common among patients with underlying hematological malignancies and those patients who require chronic transfusion support, such as SCD. However, there are few case reports of hemolytic transfusion reactions due to HLA alloimmunization Citation[19,20]. It is possible that the other host factors (i.e., activated macrophages) might also play a role in the process of RBC destruction.
In our previous six published cases, HLA antibodies were detected in four patients. Therefore, we have previously also suggested that some of the transfused RBCs expressing HLA antigens are destroyed directly by (activated macrophages) through HLA antigen–antibody immune mechanisms, and those transfused RBCs that are not expressing HLA antigens and some of the patient’s own cells might be indirectly destroyed by a HLA-mediated bystander hemolysis mechanism Citation[4,5].
Laboratory investigations
The acute form of HS poses a diagnostic challenge because identification of a new RBC antibody is often absent. A high index of suspicion is important. Laboratory investigations should include: DAT, antibody screen, close attention of reticulocyte count Citation[2], serum bilirubin level, LDH, serial measurement of RBC Hb electropheresis Citation[6] and HPLC analysis of the urine Citation[4]. This may allow early recognition Citation[6].
Management
Awareness and recognition of HS is important. Further transfusion may exacerbate ongoing hemolysis, worsen the degree of anemia and lead to a protracted course or even death Citation[1,2,9,11]. Management depends upon the severity of anemia and the speed of hemolysis.
In the mild form, additional transfusion should be avoided, and oral prednisolone (1–2 mg/kg/day) Citation[2] should be tried and the Hb level monitored closely.
If presenting with rapid severe hemolysis, the patient may require transfusion. As additional transfusion may exacerbate hemolysis, IVIG–steroid cover should be given at the same time Citation[21,22]. Use of IVIG should be selective because it has been associated with renal toxicity, thrombosis and an estimated 0.6% risk of stroke, and patients with SCD may be predisposed to such adverse events Citation[5,6].
Intravenous immunoglobulin in a low-dose regime of 0.4 g/kg/day for 5 days and intravenous methylprednisolone 0.5 g/day (adults) and 4 mg/kg (pediatric patients) for 2 days has been recommended Citation[6]. In severe hemolysis, additional IVIG–steroid therapy should be considered Citation[4,6].
Erythropoietin has been tried in HS, but further evaluation is required to substantiate its role.
Mechanism of IVIG & methylprednisolone
Intravenous immunoglobulin and methylprednisolone appear to have a synergistic effect in suppressing the activity of macrophages. Steroids have a direct suppressive action on macrophages Citation[23].
There are several possible mechanisms of IVIG on macrophages. Fever is one of the common presenting features of HS. Infection and/or fever can activate macrophages. IVIG is a pooled product that exerts a broad range of antiviral/antibacterial antibody specificity, which may neutralize infection and thus reduce the activation of macrophages Citation[24]. Animal studies have shown that IVIG prevents venular vaso-occlusion by inhibiting white cell adherence with sickle RBCs Citation[25]. Therefore, IVIG may block the adhesion of sickle RBCs and reticulocytes to macrophages. IVIG may also suppress activated macrophages through an immunomodulatory mechanism Citation[26].
Evidence is emerging that IVIG and steroid therapy may shorten the course of hemolysis and the patient might recover without requiring additional transfusion. This has been described in the severe delayed form of HS in non-SCD patients and hemolytic transfusion reaction secondary to HLA alloimmunization Citation[27,20].
A similar outcome was also observed in two of our previously reported cases Citation[5,6].
Mechanism of erythropoietin
Erythropoietin has been tried in HS Citation[6–7]; however, further evaluation is required to substantiate its role. Serum erythropoietin levels are abnormally low for the extent of anemia in SCD Citation[28]. Erythropoietin is a growth factor for both endothelial cells and erythroid cells. Trial et al. have reported that, “decreased level of erythropoietin may trigger a process called neocytolysis”. Neocytolysis is the destruction of young red cells at the endothelial–macrophage interface and the authors have speculated that this is due to “decreased production of macrophage deactivating TGF-β by endothelial cells, resulting in activation of adhesions on macrophages which triggers phagocytosis of young red cells” Citation[29]. Erythropoietin might correct anemia in HS either by directly stimulating erythroid precursors or by preventing neocytolysis resulting response in reticulocyte count . This warrants further exploration.
Recurrent HS
Hyperhemolysis syndrome may recur but this seems to be extremely rare. There are only four reported cases of recurrent episodes of HS in SCD patients – two adults and two pediatric patients. Comparative studies among different patients showed both adherence to macrophages and erythrophagocytosis to be highly variable among SCD patients Citation[30]. RBC survival studies in the same individual during two separate occasions at asymptomatic steady state demonstrated a variation in RBC destruction Citation[31]. Therefore, because of these variations, it is not possible to predict which patients may have recurrence episodes of HS after subsequent transfusion.
Hyperhemolysis syndrome is a serious and potentially life-threatening complication of RBC transfusion. The incidence of HS among patients with SCD is unknown but has now been well described. Awareness is important; without a high index of suspicion the case may go unrecognized. Treatment with IVIG–steroids should be considered in severe life-threatening HS Citation[21,22].
Financial & competing interests disclosure
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Related Research Data
References
- Friedman DF, Kim HC, Manno CS. Hyperhaemolysis associated with red cell transfusion in sickle cell disease. Transfusion33(Suppl.), 148 (1993).
- Petz LD, Calhoun L, Shulman I, Johnson C, Herron R. The sickle cell haemolytic transfusion reaction syndrome. Transfusion37, 382–392 (1997).
- Cullis JO, Win N, Dudley JM, Kaye T. Post transfusion hyperhaemolysis in a patient with sickle cell disease: use of steroids and intravenous immunoglobulin to prevent further red cell destruction. Vox Sang.4, 355–357 (1995).
- Win N, Doughty H, Telfer P, Wild B, Pearson T. Hyperhaemolytic transfusion reaction in sickle cell disease. Transfusion41, 323–328 (2001).
- Win N, Tullie Y, Needs M, Chen PE, Okpala I. Use of intravenous immunoglobulin and intravenous methylprednisolone in hyperhaemolysis syndrome in sickle cell disease. Haematology9, 433–436 (2004).
- Win N, New H, Lee E, de la Fuente J. Hyperhemolysis syndrome in sickle cell disease: case report (recurrent episode) and literature review. Transfusion48, 1231–1238 (2008).
- Talano JM, Hillery CA, Gottschall JL, Baylerian DM, Scott JP. Delayed haemolytic transfusion reaction/hyperhaemolysis syndrome in children with sickle cell disease. Pediatrics111, 661–665 (2003).
- King KE, Shirey RS, Lankjewicz MW, Young-Ramsaran J, Ness P. Delayed haemolytic transfusion reactions in sickle cell disease: simultaneous destruction of recipients’ red cells. Transfusion37, 376–381 (1997).
- Treleaven JG, Win N. Hyperhaemolysis syndrome in a patient with myelofibrosis. Haematology9, 147–149 (2004).
- Sirchia G, Moerelati F, Rebulla P. The sickle cell haemolytic transfusion reaction syndrome. Transfusion37, 1098–1099 (1997).
- Darabi K, Dzik S. Hyperhaemolysis syndrome in anaemia of chronic disease. Transfusion45, 1930–1933 (2005).
- Win N. Blood transfusion therapy for haemoglobulinopathies. In: Practical Management of Haemoglobinopathies. Okpala I (Ed.). Blackwell Publishing, Oxford, UK 99–106 (2004).
- Petz LD, Garratty G. Bystander Immune Haemolysis Immune Haemolytic Anaemias (2nd Edition). Churchill Livingstone, PA, USA 358–364 (2004).
- Garratty G. Severe reactions associated with transfusion of patients with sickle cell disease. Transfusion37, 357–361 (1997).
- Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease potential role in the activation of vascular endothelium and vaso-occlusion. Blood96, 2451–2459 (2000).
- Schwartz RS, Tanaka Y, Fidler IJ et al. Increased adherence of sickled and phosphatidylserine-enriched human erythrocytes to cultured human peripheral blood monocytes. J. Clin. Invest.75, 1965–1972 (1985).
- Gee BE, Platt OS. Sickle reticulocytes adhere to VCAM-1. Blood85, 268–274 (1995).
- Ihanus E, Uotila LM. Toivanen A, Varis M, Gahmberg CG. Red cell ICAM-4 is a ligand for the monocyte/machrophage integrin CD11c/CD18: characterization of the binding sites on ICAM-4. Blood109, 802–810 (2007).
- Takeuchi C, Ohto H, Miura S, Yasuda H, Ono S, Ogata T. Delayed and acute hemolytic transfusion reactions resulting from red cell antibodies and red cell-reactive HLA antibodies. Transfusion45, 1925–1929 (2005).
- Muro M, Palacios S, Moya-Quiles R, Candela M, Alvarez-Lopez R. Acute haemolytic transfusion reactions secondary to human leukocyte antigen alloimmunization. Int. J. Lab. Hematol.30(1), 84–86 (2008).
- Anderson D, Ali K, Blanchette V et al. Guidelines on the use of intravenous immune globulin for haematologyic conditions. Transfusion Med. Rev.21, S9–S56 (2007).
- Provan D, Nokes TJC, Agrawal S, Winer J, Wood P. Clinical Guidelines for Immunoglobulin Use. Department of Health, London, UK (2008).
- Packer JT, Greendyke RM, Swisher SN. The inhibition of erythrophagocytosis in vitro by corticosteroids. Trans. Assoc. Am. Phys.73, 93–102 (1960).
- Krause J, Wu R, Sherer Y, Patanick M, Peter JB, Shoenfeld Y. In vitro antiviral and antibacterial activity of commercial intravenous immunoglobulin preparations – a potential role for adjuvant intravenous immunoglobulin therapy in infectious disease. Transfusion Med.12, 133–139 (2002).
- Turhan A, Jenab P, Bruhns P, Ravetch JV, Coller BS, Frenette PS. Intravenous immune globulin prevents venula vaso-occlusion in sickle cell mice by inhibiting erythrocytes and adherent leuckocytes. Blood15, 2397–2399 (2004).
- Rhoades CJ, Williams MA, Kelsey SM, Newland AC. Monocyte–macrophage system as targets for immuno-modulation by intravenous immunoglobulin. Blood Rev.14, 14–30 (2000).
- Mota MA, Sakashita AM, Hamerschlak N, Kutner JM, Castilho LM. Post-transfusion hyperhaemolysis after haemolytic transfusion reaction in a patient with severe anaemia: case report. Vox Sang.91(Suppl. 3), 125; (2006) (Abstract 233).
- Sherwood JB, Goldwasser E, Chilcote R et al. Sickle cell anemia patients have low erythropoietin levels for their degree of anemia. Blood67, 46–49 (1986).
- Trial J, Rice L. Erythropoietin withdrawal leads to the destruction of young red cells at the endothelial–macrophage interface. Curr. Pharmaceut. Design10(2), 183–190 (2004).
- Hebbel RP, Miller VB. Phagocytosis of sickle erythrocyte: immunological and oxidative determinants of haemolytic anemia. Blood64, 733–741 (1984).
- Mendoza E, Gutgsell N, Temple JD, Issitt P. Monocyte phagocyte activity in sickle cell disease. Acta Haematol.85, 199–201 (1991).